Производные пиразола или их соли с основаниямиПатент на изобретение №: 2055836 Автор: Масатоси Баба[JP], Такуя Какута[JP], Норио Танака[JP], Еити Оя[JP], Такаси Икаи[JP], Тсутому Навамаки[JP], Сигеоми Ватанабе[JP] Патентообладатель: Ниссан Кемикал Индастриз Лтд. (JP) Дата публикации: 10 Марта, 1996 Адрес для переписки: подача заявки21.05.1992 публикация патента10.03.1996 Изображения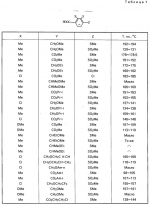 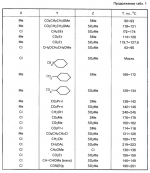 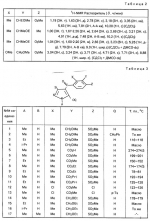 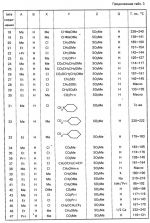 Сущность изобретения: производные пиразола ф-лы 1, где А - С1 - С3-алкил. В-водород или С1 - С3-алкил, Х - С1 - С6-алкил, хлор или C1 - C6-алкокси, У-группа ф-лы CCOR1, где R1 - водород, C1 - C6-алкил, хлор, С1 - С6-алкил, или метоксиэтил, или группа ф-лы ZOR2, где Z - метилен, возможно замещенный на метил или этил, R2 - водород, С1 - С8-алкил, 2,2,2-трифторэтил, или пропаргил, или группа ф-лы СН2С-СН2-СН2-ОR3, Z-хлор, метилсульфонил, Q-водород, финилсульфонил, бензил при соответствующих значениях радикалов, или их соли с основаниями, обладающие гербицидной активностью, особенно на суходольных полях. 6 табл. Структура соединения ф-лы 1: 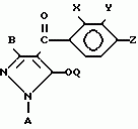 , , , , , , , , , , , , , , , , ,
, , , , , , , , , , , , , , , , , ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ПАТЕНТУИзобретение касается новых производных 4-бензоилпиразола и избирательных гербицидов, содержащих такие производные в качестве активных ингредиентов, которые особенно полезны как гербициды на суходольных полях. Известно, что некоторые производные соединения 4-бензоилпиразола обладают гербицидной активностью. Например, пиразолат (обиходное название) и пиразоксифен (обиходное название) используются, в частности, в качестве гербицидов на рисовых полях. Проявляя отличную гербицидную активность на орошаемых полях, эти соединения не годятся в качестве гербицидов на суходольных полях, поскольку их гербицидная активность низка против сорняков на суходольных полях. Среди производных 4-бензоилпиразола желательно разработать соединение, полезное в качестве гербицида на суходольных полях. Изобретение предусматривает производное пиразола, имеющее формулу 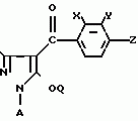 где А-(С1-С3) алкил, В-водород или (С1-С3) алкил, Х-(С1-С6) алкил, хлоро или (С1-С6) алкокси; Y группа формулы СООR1, где R1 водород, (С1-С6) алкил, хлор (С1-С6) акил или метоксиэтил или группа формулы LOR2, где L метилен, возможно замещенный на метил или этил, R2 водород, (С1 С8) алкил, 2,2,2-трифторэтил или пропаргил или группа формулы СН2-О-СН2-СН2-ОR3, где R3 (С1-С6) алкил или группа формулы СН2R4, где R4циано или S(O)nR5, где R5 означает (С1-С6) алкил; n целое число от 0 до 2 или группа формулы СН-СНOR6, где R6-(С1-С6) алкил; Z хлор или метилсульфонил; Q водород, фенилсульфонил или бензил или их соли с основаниями. Если Q является атомом водорода, то соединение может легко образовать соль с металлом или органическим основанием. В качестве таких металлов можно упомянуть натрий, калий, кальций, литий, барий, магний, железо, медь, никель или марганец. Из числа органических оснований можно упомянуть метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, н-пропиламин, ди-н-пропиламин, изо-пропиламин, диизопропиламин, н-бутиламин, изобутиламин, втор-бутиламин, третбутиламин, пиперидин, пирролидин, морфолин, пиридин, N,N-диметиланилин или холин. При проведении исследований гербицидных свойств различных органических соединений с целью получения полезных гербицидов авторы изобретения установили, что упомянутое соединение согласно настоящему изобретению проявляет высокую гербицидную активность против узколистных сорняков (травянистых и сытьевых сорняков) и против широколиственных сорняков без существенной цитотоксичности против полезных растений, т.е. культурных растений, таких как Zea mays (кукуруза), Sorghum bicolor (сорго), Triticum spp (пшеница) и Hordeum vulgare (ячмень). Настоящее изобретение было создано на основе этого открытия. Соединение согласно изобретению проявляет высокие гербицидные активности как при обработке почвы, при введении в почву, так и при обработке листьев. С другой стороны, оно не проявляет фитотоксичности к культурным растениям, таким как Zea maus, Sorghum bicolor, Triticum spp и Hordeum vulgare как при поверхностной обработке почвы, так и при введении и заделке в почву и при обработке листвы. Таким образом, соединение согласно изобретению обладает высокой избирательностью и является весьма эффективным в борьбе с сорняками в процессе выращивания этих культурных растений. А именно, соединение согласно изобретению проявляет сильные гербицидные активности против вреднейших сорняков, таких как Setaria viridis (щетинник зеленый), Echinochloa crus- galli (просо куриное). Аmaranthus lividus (ширица синеватая), Polygonum longisetum (горец грубый), Xanthium strumarium (дурнишник), аbutilon theophrasti (канатник Теофраста), Cyperus esculentus (сыть съедобная), которые развиваются при выращивании Zea mays u Sorghum bicolor. Гербицидные активности против травянистых сорняков и сыти съедобной примечательно высокие и крайне редкие. Раньше при выращивании Zea mays или Sorghum bicolor обычно применяли атразин или цианазин, как гербициды триазинового типа, или алахлор или метолахлор, как гербициды анилида кислот. Однако атразин и цианазин обладали слабыми гербицидными активностями против травянистых сорняков, хотя они проявляли высокие активности против широколиственных сорняков, и их активность против Cyperus esculentus была незначительной. С другой стороны, алахлор и метолахлор имеют слабые активности против широколиственных сорняков, хотя их активности против травянистых сорняков высокие, а их активность против Сyperus esculentus очень слабая. Таким образом было трудно искоренить все виды сорняков единственным применением этих гербицидов. В результате различных исследований было найдено соединение согласно изобретению, проявляющее высокое гербицидное действие против широкого спектра сорняков, и настоящее изобретение разработано на базе этого открытия. Соединение согласно изобретению отличается также и тем, что оно не проявляет фитотоксичности против культурных растений, таких как Zea maus, Sorghum bicolor. Triticum spp и Hordeum vulgare и таким образом может безопасно применяться на полях таких культурных растений. Далее соединение согласно изобретению включает соединение, которое показывает селективность между Oryza sativa (рисом) и Echinochloa crus-galli (просом куриным), а также включает соединение, проявляющее избирательность к полезным растениям, таким как Gossypium spp. (хлопчатник), Вeta vulgaris (сахарная свекла) или Glycine mak (соя культурная). До настоящего времени было известно, что производные 4-бензоилпиразола обладают высокой гербицидной активностью. Например, пиразолат (обиходное название) имеется в продаже и широко используется в сельскохозяйственной практике. Однако такие обычные гербициды ограничены в своем применении на заливаемых полях, и их активность очень низкая на суходольных полях. И как результат широких исследований в течение ряда лет 4-бензоилпиразольных производных было найдено, что соединение согласно настоящему изобретению, которое одновременно удовлетворяет различным условиям замещения в структуре, как отмечалось выше, проявляет высокую гербицидную активность при применении на суходольных полях, для обработки почвы, введения в почву и для обработки листьев. Было установлено, что соединение согласно изобретению обладает особенно высокой активностью против травянистых сорняков и сыти съедобной. (Сyperus esculentus). Соединение согласно изобретению может быть легко получено одной из следующих реакций. Z где А-(С1-С3) алкил, В-водород или (С1-С3) алкил, Х-(С1-С6) алкил, хлоро или (С1-С6) алкокси; Y группа формулы СООR1, где R1 водород, (С1-С6) алкил, хлор (С1-С6) акил или метоксиэтил или группа формулы LOR2, где L метилен, возможно замещенный на метил или этил, R2 водород, (С1 С8) алкил, 2,2,2-трифторэтил или пропаргил или группа формулы СН2-О-СН2-СН2-ОR3, где R3 (С1-С6) алкил или группа формулы СН2R4, где R4циано или S(O)nR5, где R5 означает (С1-С6) алкил; n целое число от 0 до 2 или группа формулы СН-СНOR6, где R6-(С1-С6) алкил; Z хлор или метилсульфонил; Q водород, фенилсульфонил или бензил или их соли с основаниями. Если Q является атомом водорода, то соединение может легко образовать соль с металлом или органическим основанием. В качестве таких металлов можно упомянуть натрий, калий, кальций, литий, барий, магний, железо, медь, никель или марганец. Из числа органических оснований можно упомянуть метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, н-пропиламин, ди-н-пропиламин, изо-пропиламин, диизопропиламин, н-бутиламин, изобутиламин, втор-бутиламин, третбутиламин, пиперидин, пирролидин, морфолин, пиридин, N,N-диметиланилин или холин. При проведении исследований гербицидных свойств различных органических соединений с целью получения полезных гербицидов авторы изобретения установили, что упомянутое соединение согласно настоящему изобретению проявляет высокую гербицидную активность против узколистных сорняков (травянистых и сытьевых сорняков) и против широколиственных сорняков без существенной цитотоксичности против полезных растений, т.е. культурных растений, таких как Zea mays (кукуруза), Sorghum bicolor (сорго), Triticum spp (пшеница) и Hordeum vulgare (ячмень). Настоящее изобретение было создано на основе этого открытия. Соединение согласно изобретению проявляет высокие гербицидные активности как при обработке почвы, при введении в почву, так и при обработке листьев. С другой стороны, оно не проявляет фитотоксичности к культурным растениям, таким как Zea maus, Sorghum bicolor, Triticum spp и Hordeum vulgare как при поверхностной обработке почвы, так и при введении и заделке в почву и при обработке листвы. Таким образом, соединение согласно изобретению обладает высокой избирательностью и является весьма эффективным в борьбе с сорняками в процессе выращивания этих культурных растений. А именно, соединение согласно изобретению проявляет сильные гербицидные активности против вреднейших сорняков, таких как Setaria viridis (щетинник зеленый), Echinochloa crus- galli (просо куриное). Аmaranthus lividus (ширица синеватая), Polygonum longisetum (горец грубый), Xanthium strumarium (дурнишник), аbutilon theophrasti (канатник Теофраста), Cyperus esculentus (сыть съедобная), которые развиваются при выращивании Zea mays u Sorghum bicolor. Гербицидные активности против травянистых сорняков и сыти съедобной примечательно высокие и крайне редкие. Раньше при выращивании Zea mays или Sorghum bicolor обычно применяли атразин или цианазин, как гербициды триазинового типа, или алахлор или метолахлор, как гербициды анилида кислот. Однако атразин и цианазин обладали слабыми гербицидными активностями против травянистых сорняков, хотя они проявляли высокие активности против широколиственных сорняков, и их активность против Cyperus esculentus была незначительной. С другой стороны, алахлор и метолахлор имеют слабые активности против широколиственных сорняков, хотя их активности против травянистых сорняков высокие, а их активность против Сyperus esculentus очень слабая. Таким образом было трудно искоренить все виды сорняков единственным применением этих гербицидов. В результате различных исследований было найдено соединение согласно изобретению, проявляющее высокое гербицидное действие против широкого спектра сорняков, и настоящее изобретение разработано на базе этого открытия. Соединение согласно изобретению отличается также и тем, что оно не проявляет фитотоксичности против культурных растений, таких как Zea maus, Sorghum bicolor. Triticum spp и Hordeum vulgare и таким образом может безопасно применяться на полях таких культурных растений. Далее соединение согласно изобретению включает соединение, которое показывает селективность между Oryza sativa (рисом) и Echinochloa crus-galli (просом куриным), а также включает соединение, проявляющее избирательность к полезным растениям, таким как Gossypium spp. (хлопчатник), Вeta vulgaris (сахарная свекла) или Glycine mak (соя культурная). До настоящего времени было известно, что производные 4-бензоилпиразола обладают высокой гербицидной активностью. Например, пиразолат (обиходное название) имеется в продаже и широко используется в сельскохозяйственной практике. Однако такие обычные гербициды ограничены в своем применении на заливаемых полях, и их активность очень низкая на суходольных полях. И как результат широких исследований в течение ряда лет 4-бензоилпиразольных производных было найдено, что соединение согласно настоящему изобретению, которое одновременно удовлетворяет различным условиям замещения в структуре, как отмечалось выше, проявляет высокую гербицидную активность при применении на суходольных полях, для обработки почвы, введения в почву и для обработки листьев. Было установлено, что соединение согласно изобретению обладает особенно высокой активностью против травянистых сорняков и сыти съедобной. (Сyperus esculentus). Соединение согласно изобретению может быть легко получено одной из следующих реакций. Z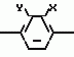 COOH + N COOH + N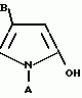  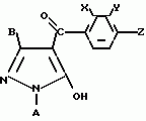 (1) Z (1) Z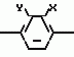 COCl + N COCl + N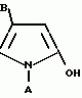 _____ _____ 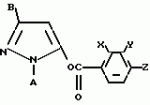  N N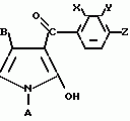 (2) (2) 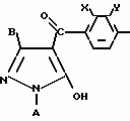 Z + E-Q Z + E-Q  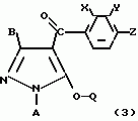 (3) (3) 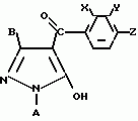 (4) (4)  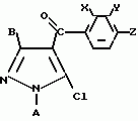  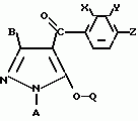 В приведенных выше формулах А,В,Х,У,Z,Q имеют значения, определенные для них ранее, Е является атомом галогена. Метансульфонилоксигруппой или пара-толуолcульфонилоксигруппой. Далее В приведенных выше формулах А,В,Х,У,Z,Q имеют значения, определенные для них ранее, Е является атомом галогена. Метансульфонилоксигруппой или пара-толуолcульфонилоксигруппой. Далее 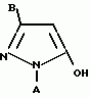 является таутомером является таутомером 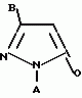 а может быть представлен той или другой формулой. ДСС означает N,N"-дициклогексилкарбодиимид. Схема реакции (1) представляет собой реакцию, в которой бензойная кислота, имеющая подходящие заместителя, и 5-оксипиразол взаимодействуют в инертном растворителе в присутствии ДСС и основания с получением 4-бензоил-5-оксипиразола. ДСС используют в количестве от 1,0 до 1,5 молей на моль бензойной кислоты и пиразола. Растворителем может быть любой растворитель, инертный в условиях реакции. Особенно предпочтительны трет-бутиловый спирт, трет-амиловый спирт или изопропиловый спирт. Основание может требоваться необязательно. Однако, в общем, выход может быть увеличен с использованием основания. Особых ограничений к основаниям нет, но карбонат калия или карбонат натрия могут быть использованы предпочтительно. Температура реакций может быть в интервале от комнатной до температуры кипения растворителя, но предпочтительно 50-100оС. Время реакции обычно составляет 0,5-20 ч. Схема реакции (2) показывает реакцию, в которой хлористый бензоил, имеющий соответствующих заместителей, взаимодействует с 5-оксипиразолом с образованием сложного бензоильного эфира, который затем подвергают перегруппировке до 4-бензоильного соединения. Бензоильная этерификация может быть осуществлена в инертном растворителе (таком как ароматический углеводород, сложный эфир жирной кислоты, галогенизированный углеводород, простой эфир, ацетонитрил, диметилсульфоксид или N, N"-диметилформамид или в двухфазной системе с таким растворителем и водой или в смеси таких растворителей в присутствии подходящего дегидрохлорирующего средства (т.е. неорганического основания, такого как гидроокись натрия, гидроокись калия или кислый карбонат натрия, или органического основания, такого как пиридин или триэтиламин) при температуре от комнатной температуры до 100оС в течение времени 10 мин 5 ч. Перегруппировка может быть проведена с помощью кислоты Льюиса, такой как безводный хлорид алюминия или основания. В качестве основания могут быть использованы карбонат калия, гидроокись кальция или карбонат натрия. Кислоту Льюиса и основание обычно используют в количестве от 1 до 10 молей. Растворитель не требуется. Однако, в некоторых случаях будет полезным использование растворителя, имеющего подходящую точку кипения для удобства в работе и увеличения выхода. Примером, подтверждающим преимущества использования растворителя, является применение диоксана или диглима. Температура реакции обычно находится в интервале 50-150оС и время реакции обычно составляет 15 мин 10 ч. Схема реакции (3) представляет реакцию, в которой 4-бензоил-5-оксипиразол конденсируют с галогенидом, сложным эфиром метансульфоновой кислоты или сложным эфиром пара-толуолсульфокислоты. Для этой реакции предпочтительно применять 1-3 молей дегидрогалогенизирующего средства. Среди дегидрогало- генизирующих средств могут быть упомянуты неорганические основания, такие как гидроокись натрия, гидроокись калия, карбонат натрия, кислый карбонат натрия или карбонат калия, или органические основания, такие как пиридин или триэтиламин. Особых ограничений к растворителям нет, коль скоро он инертен в условиях реакции. Может быть использован широкий выбор растворителей, включающий ароматический углеводород, сложный эфир жирной кислоты, галогенизированный углеводород, простой эфир, кетон, алифатический углеводород, ацетонитрил, диметилсульфоксид и диметилформамид. Температура реакции может быть необязательно выбрана в интервале от комнатной температуры до точки кипения растворителя. Время реакции обычно составляет 30 мин 30 ч. Схема реакции (4) представляет реакцию, в которой 4-бензоил-5-оксипиразол превращают в 5-хлорсоединение с помощью хлорирующего средства с последующей конденсацией с подходящим спиртом или кислотой. В качестве хлорирующего средства могут быть упомянуты хлорокись фосфора, пентахлорид фосфора или хлористый тионил. Растворителем могут служить многие инертные в условиях реакции растворители, такие как, например, диметилформамид. Температура реакции находится в интервале 30-150оС, а время реакции обычно составляет 30 мин 10 ч. В некоторых случаях время реакции может быть сокращено или увеличен выход добавлением дегидрогалогенизирующего средства. Реакцию со спиртом или кислотой проводят при добавлении дегидрогалогенизирующего средства. Дегидрогенизирующим средством могут служить основания, такие как гидроокись натрия, гидроокись калия, карбонат натрия, карбонат калия, алкоголят натрия или гидрид натрия. Может быть использован любой растворитель, инертный в условиях реакции (такой как ароматический углеводород, простой эфир, кетон или N,N"-диметилформамид). Температура реакции может быть выбрана в интервале от комнатной температуры до точки кипения растворителя. Бензойные кислоты или хлористые бензоилы, используемых в качестве исходных материалов для получения соединений согласно изобретению могут быть легко получены подходящим сочетанием известных методов синтеза. Например, соединения, в которых заместителем Z в бензольном кольце является группа S(O)n CH3, могут быть получены по следующей схеме реакций CH3 а может быть представлен той или другой формулой. ДСС означает N,N"-дициклогексилкарбодиимид. Схема реакции (1) представляет собой реакцию, в которой бензойная кислота, имеющая подходящие заместителя, и 5-оксипиразол взаимодействуют в инертном растворителе в присутствии ДСС и основания с получением 4-бензоил-5-оксипиразола. ДСС используют в количестве от 1,0 до 1,5 молей на моль бензойной кислоты и пиразола. Растворителем может быть любой растворитель, инертный в условиях реакции. Особенно предпочтительны трет-бутиловый спирт, трет-амиловый спирт или изопропиловый спирт. Основание может требоваться необязательно. Однако, в общем, выход может быть увеличен с использованием основания. Особых ограничений к основаниям нет, но карбонат калия или карбонат натрия могут быть использованы предпочтительно. Температура реакций может быть в интервале от комнатной до температуры кипения растворителя, но предпочтительно 50-100оС. Время реакции обычно составляет 0,5-20 ч. Схема реакции (2) показывает реакцию, в которой хлористый бензоил, имеющий соответствующих заместителей, взаимодействует с 5-оксипиразолом с образованием сложного бензоильного эфира, который затем подвергают перегруппировке до 4-бензоильного соединения. Бензоильная этерификация может быть осуществлена в инертном растворителе (таком как ароматический углеводород, сложный эфир жирной кислоты, галогенизированный углеводород, простой эфир, ацетонитрил, диметилсульфоксид или N, N"-диметилформамид или в двухфазной системе с таким растворителем и водой или в смеси таких растворителей в присутствии подходящего дегидрохлорирующего средства (т.е. неорганического основания, такого как гидроокись натрия, гидроокись калия или кислый карбонат натрия, или органического основания, такого как пиридин или триэтиламин) при температуре от комнатной температуры до 100оС в течение времени 10 мин 5 ч. Перегруппировка может быть проведена с помощью кислоты Льюиса, такой как безводный хлорид алюминия или основания. В качестве основания могут быть использованы карбонат калия, гидроокись кальция или карбонат натрия. Кислоту Льюиса и основание обычно используют в количестве от 1 до 10 молей. Растворитель не требуется. Однако, в некоторых случаях будет полезным использование растворителя, имеющего подходящую точку кипения для удобства в работе и увеличения выхода. Примером, подтверждающим преимущества использования растворителя, является применение диоксана или диглима. Температура реакции обычно находится в интервале 50-150оС и время реакции обычно составляет 15 мин 10 ч. Схема реакции (3) представляет реакцию, в которой 4-бензоил-5-оксипиразол конденсируют с галогенидом, сложным эфиром метансульфоновой кислоты или сложным эфиром пара-толуолсульфокислоты. Для этой реакции предпочтительно применять 1-3 молей дегидрогалогенизирующего средства. Среди дегидрогало- генизирующих средств могут быть упомянуты неорганические основания, такие как гидроокись натрия, гидроокись калия, карбонат натрия, кислый карбонат натрия или карбонат калия, или органические основания, такие как пиридин или триэтиламин. Особых ограничений к растворителям нет, коль скоро он инертен в условиях реакции. Может быть использован широкий выбор растворителей, включающий ароматический углеводород, сложный эфир жирной кислоты, галогенизированный углеводород, простой эфир, кетон, алифатический углеводород, ацетонитрил, диметилсульфоксид и диметилформамид. Температура реакции может быть необязательно выбрана в интервале от комнатной температуры до точки кипения растворителя. Время реакции обычно составляет 30 мин 30 ч. Схема реакции (4) представляет реакцию, в которой 4-бензоил-5-оксипиразол превращают в 5-хлорсоединение с помощью хлорирующего средства с последующей конденсацией с подходящим спиртом или кислотой. В качестве хлорирующего средства могут быть упомянуты хлорокись фосфора, пентахлорид фосфора или хлористый тионил. Растворителем могут служить многие инертные в условиях реакции растворители, такие как, например, диметилформамид. Температура реакции находится в интервале 30-150оС, а время реакции обычно составляет 30 мин 10 ч. В некоторых случаях время реакции может быть сокращено или увеличен выход добавлением дегидрогалогенизирующего средства. Реакцию со спиртом или кислотой проводят при добавлении дегидрогалогенизирующего средства. Дегидрогенизирующим средством могут служить основания, такие как гидроокись натрия, гидроокись калия, карбонат натрия, карбонат калия, алкоголят натрия или гидрид натрия. Может быть использован любой растворитель, инертный в условиях реакции (такой как ароматический углеводород, простой эфир, кетон или N,N"-диметилформамид). Температура реакции может быть выбрана в интервале от комнатной температуры до точки кипения растворителя. Бензойные кислоты или хлористые бензоилы, используемых в качестве исходных материалов для получения соединений согласно изобретению могут быть легко получены подходящим сочетанием известных методов синтеза. Например, соединения, в которых заместителем Z в бензольном кольце является группа S(O)n CH3, могут быть получены по следующей схеме реакций CH3 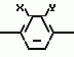 S(O)nCH S(O)nCH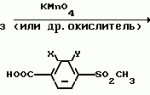 (5) (5)   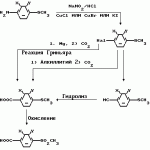  Далее получение бензойных кислот будет описано со ссылкой на ссылочные примеры. Однако следует понимать, что изобретение ни в коей мере не ограничено такими конкретными примерами. С с ы л о ч н ы й п р и м е р 1. Получение 4-метансульфонил-3-метоксиметил-2-метил- бензойной кислоты и 3-метоксиметил-2-метил-4-метилтиобензойной кислоты. 1) 2-метил-3-нитробензиловый спирт. 39,0 г (0,2 моля) метилового эфира 2-метил-3-нитробензойной кислоты растворяли в 600 мл трет-бутанола и к ним добавляли 19,0 г боргидрида натрия. В реакционную смесь при нагревании с обратным холодильником по каплям добавляли 150 мл метанола в течение 1 ч. Нагревание с обратным холодильником продолжали еще один час для завершения реакции. Реакционную смесь оставляли для охлаждения и затем в нее добавляли воду. Растворитель отгоняли при пониженном давлении. К остатку добавляли воду и хлороформ, органический слой отделяли и сушили над безводным сульфатом натрия. Затем отгоняли растворитель и получали 30,7 г 2-метил-3-нитробензилового спирта. 2) 2-метил-3-нитробензилметиловый эфир. 30,1 г (0,18 моля) 2-метил-3-нитробензилового спирта, полученного на предыдущей стадии, растворяли в 200 мл бензола и в раствор последовательно добавляли 0,2 г бромистого тетра-н-бутиламмония и 20,1 г 50%-ного водного раствора гидроокиси натрия. Затем при комнатной температуре по каплям добавляли 27,2 г диметилсульфата. Дальше реакцию проводили 3 ч при перемешивании. К реакционному раствору добавляли воду и органический слой отделяли, последовательно промывали водой, 2%-ным водным раствором хлористоводородной кислоты, водой и насыщенным водным раствором хлористого натрия. Затем растворитель отгоняли и получали 30,9 г 2-метил-3-нитробензилметилового эфира в виде маслянистого вещества. 3) 3-метоксиметил-2-метиланилин. К 30,7 г (0,17 моля) полученного выше 2-метил-3-нитробензилметилового эфира добавляли 200 мл метанола. После растворения соединения в метаноле в него постепенно добавляли 92 мл концентрированной хлористоводородной кислоты. Затем в реакционную смесь постепенно добавляли 30,4 г железного порошка так, чтобы температура реакции не превышала 60оС и реакцию продолжали еще 1 ч. К реакционному раствору добавляли 300 мл воды и добавляли гидроокись натрия до тех пор, пока рН не станет более 8. К полученному шламму добавляли хлороформ и смесь тщательно перемешивали. Затем твердое вещество отделяли фильтрацией и органический слой отделяли от фильтрата. Органический слой последовательно промывали водой и насыщенным водным раствором хлористого натрия и затем сушили над безводным сульфатом натрия. Далее растворитель отгоняли при пониженном давлении и получали 23,1 г 3-метоксиметил-2-анилина в виде маслянистого вещества. 4) 3-метоксиметил-2-метил-4-тиоцианоанилин. 22,6 г (0,15 моля) 3-метоксиметил-2-метиланилина растворяли в 300 мл метанола. Затем в раствор добавляли 36,5 г тиоцианата натрия для получения однородного раствора. Этот раствор охлаждали до 0оС и в него по каплям добавляли 100 мл насыщенного метанольного раствора бромистого натрия с 25,2 г брома так, чтобы температура реакции не превышала 5оС. После капельной добавки смесь перемешивали при температуре, не превышающей 5оС 1 ч и при комнатной температуре один час для завершения реакции. Реакционный раствор выливали в 1 л воды и нейтрализовали 5%-ным водным раствором карбоната натрия. Добавляли хлороформ для экстрагирования маслянистого вещества. Хлороформовый слой промывали водой и насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении и получали 29,6 г желаемого продукта. 5) 3-метоксиметил-2-метил-4-метилтиоанилин. 29,1 г (0,14 моля) 3-метоксиметил-2-метил-4-тиоцианоанилина растворяли в 200 мл этанола и смешивали со 100 мл водного раствора, содержащего 33,6 г нонагидрата сульфата натрия при комнатной температуре. Затем в полученный раствор по каплям добавляли 21,9 г иодистого метила и смесь оставляли на 3 ч при комнатной температуре для продолжения реакции. После завершения реакции растворитель отгоняли при пониженном давлении и к остатку добавляли воду и хлороформ. Затем отделяли органический слой и последовательно промывали водой и насыщенным водным раствором хлористого натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и получали 25,6 г желаемого продукта в виде маслянистого вещества. 6) 3"-иодо-2"-метил-6"-диметилтиобензилметиловый эфир. К 25,6 г (0,13 моля) 3-метоксиметил-2-метил-4-метилтиоанилина добавляли 100 мл воды и 33 мл концентрированной хлористоводородной кислоты для превращения его в гидрохлорид анилина. Этот раствор охлаждали до 0оС и в него по каплям добавляли 30 мл водного раствора, содержащего 9,3 г нитрита натрия так, чтобы температура реакции не превышала 5оС. После завершения капельной добавки перемешивание продолжали еще 30 мин для завершения диазотирования. 100 мл водного раствора, содержащего 33 г иодистого калия, нагревали до 70оС и водный раствор соли диазония, полученного выше, постепенно добавляли к нему и разлагали. Реакционный раствор далее перемешивали 1 ч при 70оС и затем оставляли для охлаждения. Масляный компонент экстрагировали бензолом. Бензольный слой промывали последовательно водой, насыщенным раствором кислого сульфита натрия, водой и насыщенным раствором хлористого натрия в воде. Затем растворитель отгоняли при пониженном давлении и остаток очищали колоночной хроматографией (элюентом служил бензол). Получали 30,0 г желаемого продукта, точка плавления (т.пл.) 56-59оС. 7) 3-метоксиметил-2-метил-4-метилтиобензойная кислота. 27,7 г (0,09 моля) 3"-иодо-2"-метил-6"-метилтиобензилметилового эфира растворяли в 100 мл осушенного тетрагидрофурана и к раствору по каплям добавляли при -70оС 63 мл 1,5 М н-бутиллития в н-гексане. По окончании капельной добавки смесь перемешивали 15 мин при той же температуре и через реакционный раствор осторожно продували газообразную двуокись углерода до прекращения выделения тепла реакционным раствором. По окончании реакции температуру раствора доводили до комнатной температуры и добавляли воду и диэтиловый эфир для жидкостного разделения. Полученный водный слой промывали далее дважды диэтиловым эфиром, а затем концентрированной хлористоводородной кислотой устанавливали рН 1. Выпавшие в осадок кристаллы собирали фильтрацией, тщательно промывали водой, сушили и получали 14,4 г желаемого продукта. Т.пл. 192,0-194,0оС. 8) 4-метансульфонил-3-метоксиметил-2-метилбензойная кислота. К 11,3 г (0,05 моля) 3-метоксиметил-2-метил-4-метилтиобензойной кислоты добавляли 120 мл уксусной кислоты и 120 мл 35%-ного водного раствора перекиси водорода и смесь подвергали реакции при 80оС в течение 1 ч. После охлаждения реакционный раствор выливали в воду со льдом, после чего выпавшие в осадок кристаллы собирали фильтрацией, промывали водой, сушили и получали 12,3 г целевого соединения. Т.пл. 129,0-131,0оС. С с ы л о ч н ы й п р и м е р 2. Получение 3-метоксикарбонил-2-метил-4-метилтиобен- зойной кислоты и 4-метансульфонил-3-метоксикарбонил-2-метил-бензойной кислоты. 1) Метиловый эфир 3-амино-2-метилбензойной кислоты. 40 г метилового эфира 2-метил-3-нитробензойной кислоты растворяли в 120 мл метанола и в раствор добавляли 157 г концентрированной хлористоводородной кислоты. Затем добавляли постепенно 36,8 г железного порошка, поддерживая температуру не выше 60оС. Смесь перемешивали при комнатной температуре 4 ч и затем выливали в 1 л воды с льдом. Раствор нейтрализовали карбонатом натрия и экстрагировали хлороформом (после отфильтровывания нерастворимой фракции). Экстракт промывали насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 27,8 г желаемого соединения в виде маслянистого вещества. 2) Метиловый эфир 3-амино-2-метил-6-тиоцианобензойной кислоты. К раствору, содержащему 27,7 г метилового эфира 3-амино-2-метилбензойной кислоты, 41,5 г тиоцианата натрия и 250 мл метанола при температуре не выше 0оС по каплям медленно добавляли 100 мл насыщенного бромистым натрием метанола с 28,1 г бpома. Смесь перемешивали при комнатной температуре три часа и затем выливали в 1 л воды с льдом. Раствор нейтрализовали карбонатом натрия и экстрагировали хлороформом. Экстракт промывали насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 34,0 г желаемого продукта в виде маслянистого вещества. 3) Метиловый эфир 3-амино-2-метил-6-метилтиобензойной кислоты. К раствору, содержащему 39,5 г нонагидрата сульфида натрия и 110 мл воды по каплям добавляли раствор, содержащий 32,9 г метилового эфира 3-амино-2-метил-6-тиоцианобензойной кислоты и 300 мл этанола. Смесь перемешивали при комнатной температуре 1,5 ч и к нему по каплям добавляли 24,0 г иодистого метила при охлаждении льдом. Смесь перемешивали далее при комнатной температуре еще 2 ч и затем концентрировали при пониженном давлении. К ней добавляли насыщенный водный раствор хлористого натрия и смесь экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Затем отгоняли растворитель и получали 30,1 г желаемого соединения в виде маслянистого вещества. 4) Метиловый эфир 3-иодо-2-метил-6-метилтиобензойной кислоты. 28 г метилового эфира 3-амино-2-метил-6-метилтиобензойной кислоты перемешивали в 150 мл концентрированной хлористоводородной кислоты при комнатной температуре в течение 2 ч, чтобы превратить ее в гидрохлорид. Затем, поддерживая температуру смеси не выше 0оС, к ней по каплям добавляли раствор, содержащий 11,9 г нитрита натрия и 20 мл воды, чтобы получить раствор соли диазония. Раствор соли диазония по каплям добавляли к раствору, содержащему 28,4 г иодистого калия и 90 мл воды, поддерживая температуру раствора 80оС. По окончании капельного введения смесь перемешивали при 80оС в течение 15 мин и оставляли для охлаждения. В нее добавляли воду и смесь экстрагировали хлороформом. Экстракт промывали водным раствором кислого сульфита натрия и водой, затем сушили над безводным сульфатом натрия. Растворитель отгоняли и получали 40 г желаемого соединения в виде сырого продукта. Сырой продукт подвергали очистке колоночной хроматографией на силикагеле (элюируя бензолом) и получали 36,0 г очищенного соединения в виде маслянистого вещества. 5) 3-метоксикарбонил-2-метил-4-метилтиобензойная кислота. Поддерживая температуру раствора, содержащего 20,0 г метилового эфира 3-иодо-2-метил-6-метилтиобензойной кислоты и 70 мл осушенного тетрагидрофурана, не выше -60оС в атмосфере азота, к нему по каплям добавляли 42 мл 1,5 М раствора н-бутиллития в н-гексане. Через 15 мин в смесь вдували осушенный углекислый газ, поддерживая температуру не выше -50оС. После продувки азотом с целью очистки от газообразной двуокиси углерода в смесь по каплям добавляли 12,7 г диизопропиламина и смесь перемешивали до достижения комнатной температуры. Смесь концентрировали при пониженном давлении. В нее добавляли воду и промывали смесь хлороформом. Водный раствор подкисляли концентрированной хлористоводородной кислотой и затем экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Растворитель отгоняли и получали 7,5 г желаемого соединения. Т.пл. 178-178,5оС. 6) 4-метансульфонил-3-метоксикарбонил-2-метилбензойная кислота. Раствор, содержащий 5,0 г 3-метоксикарбонил-2-метил-4-метилтиобензойной кислоты, 25 мл уксусной кислоты и 25 мл 35%-ной перекиси водорода, перемешивали при 80оС 3 ч. После охлаждения смесь выливали в воду с льдом и экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 5,1 г желаемого соединения. Т.пл. 151-152оС. С с ы л о ч н ы й п р и м е р 3. Получение 2-хлор-3-этилтиометил-4-метансульфонил-бензойной кислоты. 1) Метиловый эфир 3-бромметил-2-хлор-4-метансульфонилбензойной кислоты. 12,1 г метилового эфира 2-хлор-4-метансульфонил-3-метилбензойной кислоты растворяли в 250 мл четыреххлористого углерода и раствор нагревали с обратным холодильником при перемешивании. Затем в него постепенно добавляли 7,5 г брома и 1,0 г перекиси бензоила в течение 30 мин и далее раствор нагревали с обратным холодильником 4 ч. После охлаждения в него добавляли 200 мл хлороформа и смесь промывали 5%-ным водным раствором кислого сульфита натрия. Органический слой отделяли и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и получали неочищенный продукт. Неочищенный продукт промывали этиловым эфиром и получали 13,2 г кристаллов желаемого соединения. Т.пл. 77-78оС. 2) Метиловый эфир 2-хлор-3-этилтиометил-4-метансульфонилбензойной кислоты. К 100 мл тетрагидрофурана добавляли 1,3 г этантиола и 1,5 г карбоната калия и затем 4,4 г метилового эфира 3-бромметил-2-хлор-4-метансульфонилбензойной кислоты и смесь перемешивали один день при комнатной температуре. Затем смесь перемешивали еще 1 ч при температуре 50-60оС. После охлаждения в нее добавляли хлороформ и смесь промывали разбавленным водным раствором карбоната калия. Хлороформовый слой отделяли и сушили. Затем растворитель отгоняли и получали 4,1 г метилового эфира 2-хлор-3-этилтиометил-4-метансульфонилбензойной кислоты в виде маслянистого вещества. 3) 2-хлор-3-этилтиометил-4-метансульфонилбензойная кислота. К раствору смеси, содержащему 50 мл 10%-ного водного раствора гидроокиси натрия и 150 мл метанола, добавляли 3,9 г метилового эфира 2-хлор-3-этилтиометил-4-метансульфонилбензойной кислоты и смесь перемешивали при комнатной температуре 30 мин. Метанол отгоняли при пониженном давлении и к остатку добавляли разбавленную хлористоводородную кислоту для преципитации кислоты. Смесь экстрагировали этилацетатом и экстракт сушили. Затем растворитель отгоняли и получали 3,5 г желаемого соединения. Т.пл. 172-174оС. С с ы л о ч н ы й п р и м е р 4. Получение 2-хлор-4-метансульфонил-3-метоксиметил- бензойной кислоты. 1) Метиловый эфир 2-хлор-4-метансульфонил-3-метоксиметилбензойной кислоты. К раствору, содержащему 12,0 г метилового эфира 3-бромметил-2-хлор-4-метан-сульфонилбензойной кислоты, полученному по ссылочному примеру 3 (1), и 100 мл метанола добавляли 50 мл метанолового раствора, содержащего 1,7 г метилата натрия и смесь перемешивали при комнатной температуре, оставляя на ночь. Растворитель отгоняли при пониженном давлении. Затем добавляли к остатку разбавленную хлористоводородную кислоту и смесь экстрагировали хлороформом. Экстракт промывали водой и сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 9,5 г неочищенного продукта. Неочищенное желаемое соединение очищали хроматографией на колонке силикагеля (элюировали бензолом) и получали 7,5 г очищенного желаемого соединения в виде маслянистого вещества. 2) 2-хлор-4-метансульфонил-3-метоксиметилбензойная кислота. К раствору, содержащему 3,0 г метилового эфира 2-хлор-4-метансульфонил-3-метоксиметилбензольной кислоты и 20 м метанола, добавляли раствор, содержащий 0,57 г 93%-ной гидроокиси натрия и 2 мл воды и смесь перемешивали при комнатной температуре 30 мин. После добавления 10 мл воды смесь концентрировали при пониженном давлении. Затем в нее добавляли разбавленной хлористоводородной кислоты и смесь экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 2,6 г целевого соединения. Т.пл. 137-141оС. С с ы л о ч н ы й п р и м е р 5. Получение 2-хлор-4-метансульфонил-3-метоксиметил- бензойной кислоты (альтернативный метод способу, описанному в ссылочном примере 4). Желаемое соединение получали таким же образом, что и в ссылочном примере 1. Т.пл. 137-141оС. Физические свойства промежуточных соединений следующие: 2-хлор-3-нитробензиловый спирт: масляное вещество; 2"-хлор-3"-нитробензилметиловый эфир: маслянистое вещество; 2-хлор-3-метоксиметиланилин: маслянистое вещество; 2-хлор-3-метоксиметил-4-тиоцианоани- лин: т.пл. 90-96оС; 2-хлор-3-метоксиметил-4-метилтиоани- лин: маслянистое вещество; 2"-хлор-3"-иодо-6"-метилтиобензилме-тиловый эфир, т.пл. 53-56оС. С с ы л о ч н ы й п р и м е р 6. Получение 2-хлор-4-метансульфонил-3-метоксикарбо- нилбензойной кислоты. Желаемое соединение получали таким же образом, что и в ссылочном примере 2. Т.пл. 160-162оС. Физические свойства промежуточных соединений следующие: метиловый эфир 3-амино-2-хлорбензойной кислоты, маслянистое вещество; метиловый эфир 3-амино-2-хлор-6-тиоцианобензойной кислоты, т.пл. 80-83оС; метиловый эфир 3-амино-2-хлор-6-метилтиобензойной кислоты, т.пл. 70-72оС; метиловый эфир 2-хлор-3-иод-6-метилтиобензойной кислоты, маслянистое вещество; 2-хлор-3-метоксикарбонил-4-метилтио- бензойная кислота, т.пл. 176-179оС. С с ы л о ч н ы й п р и м е р 7. Получение 4-метансульфонил-3-/(2-метоксиэтил)-окси- карбонил/-2-метилбензойной кислоты. Желаемое соединение было получено таким же образом, что и в ссылочном примере 2. Т.пл. 118-121оС. Физические свойства промежуточных соединений следующие: 2-метоксиэтиловый эфир 3-амино-2-метилбензойной кислоты, маслянистое вещество; 2-метоксиэтиловый эфир 3-амино-2-метил-6-тиоцианобензойной кислоты, т. пл.79-81оС; 2-метоксиэтиловый эфир 3-амино-2-метил-6-метилтиобензойной кислоты, маслянистое вещество; 2-метоксиэтиловый эфир 3-иодо-2-метил-6-метилтиобензойной кислоты, маслянистое вещество; 3-/(2-метоксиэтил)оксикарбонил/-2-ме- ти-4-метилтиобензойзная кислота, т.пл. 90-93оС. С с ы л о ч н ы й п р и м е р 8. Получение 2-метил-4-метилтио-З-н-пропоксикарбонилбен- зойной кислоты и 4-метансульфонил-2-метил-3-н-пропоксикарбонилбензойной кислоты 1) Метиловый эфир 3-бром-2-метил-6-метилтиобензойной кислоты. 16,1 г соединения, полученного в ссылочном примере 2 (3), перемешивали в 150 мл бромистоводородной кислоты (48%) для превращения его в бромгидрат. Поддерживая температуру не выше, чем 0оС, к полученному раствору по каплям добавляли раствор, содержащий 7,2 г нитрита натрия и 20 мл воды для получения раствора соли диазония. Раствор соли диазония по каплям добавляли к раствору, содержащему 6,0 г бромида меди и 7,7 г 48%-ной бромистоводородной кислоты, нагревая с обратным холодильником. После завершения капельной добавки смесь нагревали с обратным холодильником еще 1 ч и оставляли для охлаждения. В нее добавляли воду с льдом и смесь экстрагировали хлороформом. Экстракт промывали водным раствором кислого сульфита натрия и водой и затем сушили над безводным сульфатом натрия. Растворитель отгоняли и получали 19,2 г желаемого соединения в виде неочищенного продукта. Неочищенный продукт очищали хроматографией на колонке силикагеля (проявляли бензолом) и получали 17,1 г чистого продукта в виде маслянистого вещества. 2) 3-бром-2-метил-6-метилтиобензойная кислота. К 100 мл этанолового раствора, содержащего 17,0 г метилового эфира 3-бром-2-метил-6-метилтиобензойной кислоты, добавляли 16 г 50%-ного водного раствора гидроокиси натрия и смесь нагревали с обратным холодильником 3 ч. Реакционную смесь концентрировали при пониженном давлении. Затем в нее добавляли воду и смесь промывали хлороформом. Водный слой подкисляли концентрированной хлористоводородной кислотой и экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Растворитель отгоняли и получали 15,9 г желаемого соединения. Т.пл. 98-103оС. 3) н-пропиловый эфир 3-бром-2-метил-6-метилтиобензойной кислоты. Хлористый тионил добавляли к 15,8 г 3-бром-2-метил-6-метилтиобензойной кислоты и смесь нагревали с обратным холодильником 4 ч. Хлористый тионил отгоняли и к остатку добавляли 70 мл н-пропанола при охлаждении льдом. Затем добавляли по каплям раствор, содержащий 7,3 г пиридина и 20 мл н-пропанола. Смесь оставляли на ночь при перемешивании при комнатной температуре и концентрировали при пониженном давлении. Затем добавляли этилацетат и смесь последовательно промывали 5% -ным водным раствором карбоната натрия, 10%-ной хлористоводородной кислотой и водой и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и получали 18,0 г желаемого соединения в виде неочищенного продукта. Сырой продукт очищали хроматографией на колонке силикагеля (элюировали бензолом) и получали 16,6 г очищенного продукта в виде маслянистого вещества. 4) 3-бром-2-метил-6-метилтиобензойная кислота. Это соединение получали таким же образом, что и в ссылочном примере 2 (5). Т.пл. 138-142оС. 5) 3-бром-6-метансульфонил-2-метилбензойная кислота. Это соединение получали таким же образом, что и в ссылочном примере 2 (6). Т.пл. 142-146оС. С с ы л о ч н ы й п р и м е р 9. Получение 2-хлор-3-изопропоксикарбонил-4-метансуль- фонилбензойной кислоты. Это соединение получали из соединения по ссылочному примеру 6 (4) таким же образом, что и в ссылочном примере 8 (2). Т.пл. 146-148оС. Физические свойства промежуточных соединений следующие: 2-хлор-3-иодо-6-метилтиобензойная кислота, т.пл. 155-159оС; изопропиловый эфир 2-хлор-3-иодо-6-метилтиобензойной кислоты, маслянистое вещество; 2-хлор-3-изопропоксикарбонил-4-ме- тилтиобензойная кислота, т.пл. 114-118оС. С с ы л о ч н ы й п р и м е р 10. Получение 3-(1-метоксиэтил)-2-метил-4-метилтиобен-зойной кислоты и 4-метансульфонил-3-(1-метоксиэтил)-2-метилбензойной кислоты. 1) 2"-метил-3"-нитроацетофенон. К 5,4 г металлического магния добавляли по каплям 5 мл абсолютного этанола и 0,5 мл четыреххлористого углерода в потоке сухого азота. Далее добавляли 130 мл сухого диэтилового эфира при нагревании с обратным холодильником и затем по каплям добавляли раствор, содержащий 25 мл диэтилового эфира, 35,2 г диэтилового эфира малоновой кислоты и 20 мл этанола, со скоростью, поддерживающей температуру образования флегмы. После завершения капельного введения нагревание с обратным холодильником продолжали 3 ч для получения диэтилового эфира этоксимагнезиомалоновой кислоты. К полученному таким образом раствоpу диэтилового эфира этоксимагнезиомалоновой кислоты по каплям добавляли 150 мл раствора диэтилового эфира, содержащего 40 г хлорида 2-метил-3-нитробензойной кислоты, полученного из 2-метил-3-нитробензойной кислоты и хлористого тионила, в течение 20 мин, при нагревании с обратным холодильником и реакцию продолжали 2 ч. После охлаждения добавляли разбавленную серную кислоту для проведения гидролиза. Слой диэтилового эфира последовательно промывали водой и насыщенным водным раствором хлористого натрия. Затем растворитель отгоняли при пониженном давлении, остаток сушили и получали неочищенное соединение диэтиловый эфир 2-(2-метил-3-нитробензоил)-малоновой кислоты. В этот неочищенный продукт добавляли смесь, содержащую 7,5 мл концентрированной серной кислоты, 60 мл уксусной кислоты и 40 мл воды и смесь нагревали с обратным холодильником 6 ч. Затем рН смеси устанавливали равным 10 с помощью 20%-ного раствора гидроокиси натрия в воде. Выпавший в осадок масляный компонент экстрагировали хлороформом. Хлороформовый слой последовательно промывали водой и водным раствором хлористого натрия. Затем растворитель отгоняли при пониженном давлении и получали 34,0 г желаемого продукта. (Выход 95%), т.пл. 53-54оС. 2) Получение 1-метил-2-метил-3-нитробензилового спирта. К 50 мл метанолового раствора 0,5 г гидроокиси натрия добавляли при 0оС 0,9 г боргидрида натрия и затем к нему по каплям добавляли 100 мл метанолового раствора 14,3 г 2-метил-3-нитроацетофенона. Температуру смеси повышали до комнатной температуры и реакцию продолжали 1 ч. По завершении реакции реакционную смесь выливали в воду и экстрагировали бензолом. Последующую операцию проводили обычным образом и получали 14,3 г желаемого продукта в виде маслянистого вещества (выход 99%). В дальнейшем проводили синтез таким же образом, что и в ссылочном примере 1, для получения промежуточных соединений (3) (9). 3) 1-метил-2"-метил-3"-нитробензилметиловый эфир, маслянистое вещество. 4) 1-метил-3"-амино-2"-метилбензилметиловый эфир, маслянистое вещество. 5) 1-метил-3-амино-2-метил-6-тиоцианобензилметиловый эфир, твердое вещество. 6) 1-метил-3"-амино-2"-метил-6"-метилтиобензилметиловый эфир, маслянистое вещество. 7) 1-метил-3"-иодо-2"-метил-6"-метилтиобензил, маслянистое вещество. 8) 3-(1-метоксиэтил)-2-метил-4-метилтиобензойная кислота, маслянистое вещество, 9) 4-метансульфонил-3-(1-метоксиэтил)-2-метилбензойная кислота, т.пл. 106-109оС. С с ы л о ч н ы й п р и м е р 11. Получение 2,4-дихлор-3-метоксикарбонилбензойной кислоты. 1) 2,4-дихлор-3-нитробензойная кислота. К раствору 25 мл дымящей азотной кислоты и 20 мл серной кислоты постепенно добавляли 25 г 2,4-дихлорбензойной кислоты. После прекращения выделения тепла реакционную смесь выливали в воду с льдом. Твердый осадок промывали водой, сушили и получали 23,0 г желаемого соединения. 2) Метиловый эфир 2,4-дихлор-3-нитробензойной кислоты. 23,0 г 2,4-дихлор-3-нитробензойной кислоты и 150 мл хлористого тионила нагревали с обратным холодильником 6 ч. Затем хлористый тионил отгоняли и получали неочищенный 2,4-дихлор-3-нитробензоилхлорид. К неочищенному соединению добавляли 200 мл метанола и нагревали с обратным холодильником. Метанол отгоняли и добавляли этилацетат для получения этилацетатного раствора. Раствор последовательно промывали 5%-ным водным раствором гидроокиси натрия, разбавленной хлористоводородной кислотой и водой. После осушения растворитель отгоняли и получали 21,8 г желаемого продукта. Т.пл. 72-74оС. В дальнейшем синтез был проведен таким же образом, что и в ссылочном примере 1 для получения промежуточных соединений (3) и (4) и целевого продукта (5). 3) Метиловый эфир 3-амино-2,4-дихлорбензойной кислоты, маслянистое вещество. 4) Метиловый эфир 2,4-дихлор-3-иодобензойной кислоты, маслянистое вещество. 5) 2,4-дихлор-3-метоксикарбонилбензойная кислота, т.пл. 183-185оС. С с ы л о ч н ы й п р и м е р 12. Получение 2-хлор-3-цианометил-4-метансульфонилбен- зойной кислоты. 1) Метиловый эфир 2-хлор-3-цианометил-4-метансульфонилбензойной кислоты. 5,0 г метилового эфира 3-бромметил-2-хлор-4-метансульфонилбензойной кислоты добавляли к раствору 0,4 г 18-крауна-6 и 1,9 г цианистого калия в 50 мл ацетонитрила. Смесь перемешивали 72 ч при комнатной температуре. После отфильтровывания твердой фазы к фильтрату добавляли воду и смесь экстрагировали хлороформом. После промывки экстракта водой и сушки растворитель отгоняли и получали неочищенный продукт. Сырой продукт очищали хроматографией на короткой колонке силикагеля (элюировали хлороформом) и получали 4,1 г желаемого соединения. Т.пл. 151-155оС. 2) 2-хлор-3-цианометил-4-метансульфонилбензойная кислота. К 4,0 г метилового эфира 2-хлор-3-цианометил-4-метансульфонилбензойной кислоты и 50 мл метанола постепенно добавляли 5 мл водного раствора, содержащего 0,72 г 93% -ной гидроокиси натрия. Смесь перемешивали 15 мин при комнатной температуре. Затем реакционную смесь нейтрализовали разбавленной хлористоводородной кислотой, метанол отгоняли при пониженном давлении и концентрированный раствор экстрагировали хлороформом. После промывки экстракта водой и сушки его, хлороформ отгоняли и получали 0,9 г желаемого соединения, т.пл. 169-172оС. С с ы л о ч н ы й п р и м е р 13. Получение 3-ацетоксиметил-2-хлор-4-метансульфонил- бензойной кислоты. 1) Метиловый эфир 3-ацетоксиметил-2-хлор-4-метансульфонилбензойной кислоты. 50 мл раствора диметилформамида, содержащего 5,0 г метилового эфира 3-бромметил-2-хлор-4-метансульфонилбензойной кислоты и 1,2 г ацетата натрия, перемешивали 2 ч при 100оС. После охлаждения реакционную смесь выливали в воду с льдом и экстрагировали хлороформом. После промывки экстракта водой и сушки растворитель отгоняли и получали 4,2 г желаемого соединения. Т.пл. 165-168оС. 2) 2-хлор-3-оксиметил-4-метансульфонилбензойная кислота. 6 мл водного раствора, содержащего 1,3 г 93%-ной гидроокиси натрия, добавляли к 3,9 г метилового эфира 3-ацетоксиметил-2-хлор-4-метансульфонилбензойной кислоты в 100 мл метанола. Смесь перемешивали 30 мин при комнатной температуре. В нее добавляли 50 мл воды и метанол отгоняли при пониженном давлении. Затем реакционную смесь подкисляли хлористоводородной кислотой и экстрагировали хлороформом. Экстракт концентрировали досуха и получали 1,3 г желаемого соединения. Т.пл. 240-245оС. 3) 3-ацетоксиметил-2-хлор-4-метансульфонилбензойная кислота. 1,3 г 2-хлор-3-оксиметил-4-метансульфонилбензойной кислоты и 30 мл уксусного ангидрида нагревали с обратным холодильником 3 ч. Реакционную смесь концентрировали при пониженном давлении. Затем добавляли 50 мл воды и нагревали 1 ч. Выпавшее в осадок твердое вещество собирали фильтрацией, промывали водой и сушили. Получали 1,35 г желаемого продукта, т.пл. 219-233оС. С с ы л о ч н ы й п р и м е р 14. Получение 2,4-дихлор-3-метоксиметилбензойной кислоты. Это соединение получали таким же способом, что и в ссылочном примере 3 (1) и в примере 4. Т.пл. 130-136оС. Физические свойства промежуточных соединений следующие: 1) метиловый эфир 3-бромметил-2,4-дихлорбензойной кислоты, т.пл. 55-58оС, 2) метиловый эфир 2,4-дихлор-3-метоксиметилбензойной кислоты, маслянистое вещество. Физические свойства бензойных кислот, полученных в соответствии с предыдущими ссылочными примерами, представлены в табл.1 и 2, включающих кислоты, полученные по предшествующим ссылочным примерам. Эти бензойные кислоты легко могут быть переведены в бензоилхлориды хлорирующими средствами, такими как пятихлористый фосфор, хлористый тионил и хлористый сульфурил. При использовании таких бензойных кислот или бензоилхлоридов соединения согласно изобретению могут быть легко получены в соответствии со схемой реакций (1)-(4). Изобретение будет далее подробно описано со ссылкой на примеры. Однако следует понимать, что изобретение ни в коей мере не ограничивается такими специфическими примерами осуществления. П р и м е р 1. Получение 1-этил-5-окси-4-(4-метансульфонил-3-метоксиметил-2-ме- тилбензоил)-пиразола. 1,12 г (0,01 моля) 1-этил-5-оксипиразола растворяли в 30 мл трет-амилового спирта и в раствор последовательно добавляли 2,59 г (0,01 моля) 4-метансульфонил-3-метоксиметил-2-метилбензойной кислоты, 2,06 г (0,01 моля) N, N"-дициклогексилкарбодиимида и 0,69 г (0,005 моля) безводного карбоната калия. Реакцию в смеси проводили при температуре 80-90оС при перемешивании в течение 8 ч. После завершения реакции трет-амиловый спирт отгоняли при пониженном давлении и к остатку добавляли 30 мл воды для растворения растворимых компонентов. Смесь подвергали фильтрации для отделения нерастворимых фракций. Полученный водный раствор промывали хлороформом и добавляли концентрированную соляную кислоту, чтобы установить рН 1. Выпавший в осадок масляный компонент экстрагировали хлороформом. Растворитель отгоняли при пониженном давлении и остаток очищали хроматографией на колонке силикагеля (элюировали смесью этилацетат-этанол в отношении 9:1) и получали 2,3 г целевого продукта (выход 66% т.пл. 116-118оС). П р и м е р 2. Получение 1-этил-5-окси-4-(4-метансульфонил-3-метоксикарбонил-2-метилбензоил)-пиразола 1,12 г (0,01 моля) 1-этил-5-оксипиразола растворяли в 30 мл трет-амилового спирта и к полученному раствору последовательно добавляли 2,72 г (0,01 моля) 4-метансульфонил-3-метоксикарбонил-2-метилбензойной кислоты, 2,27 г (0,11 моля) N,N"-дициклогексилкарбонимида и 0,76 г (0,0055 моля) безводного карбоната калия. Смесь реагировала при 80оС в течение 6 ч при перемешивании. После завершения реакции трет-амиловый спирт отгоняли при пониженном давлении и затем к остатку добавляли воду для растворения растворимых компонентов. Смесь подвергали фильтрации для отделения нерастворимых веществ. Полученный водный раствор два раза промывали хлороформом и затем добавляли концентрированную хлористоводородную кислоту для установления значения рН менее 1. Выпавший в осадок масляный компонент экстрагировали хлороформом. Хлороформовый слой последовательно промывали водой и насыщенным водным раствором хлористого натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и полученный остаток перекристаллизовывали из смеси вода-этанол и получали 2,26 г целевого соединения (выход 62% т.пл. 150-152оС). П р и м е р 3. Получение 5-окси-(3-изопропоксикарбонил-4-метансульфонил-2-ме- тилбензоил)-1-метилпиразола. Операция и обработка были выполнены таким же образом, что и в примере 1, за исключением того, что 1,12 г 1-этил-5-оксипиразола заменили 0,98 г 5-окси-1-метилпиразола и 2,72 г 4-метансульфонил- 3-метоксикарбонил-2-метилбензойной кислоты были заменены 3,0 г 3-изопропоксикарбонил-4-метансульфонил-2-метилбен- зойной кислоты, и получали 1,71 г целевого соединения (выход 45% т.пл. 192-194оС). П р и м е р 4. Получение 4-(2-хлор-3-этилтиометил-4-метансульфонилбензоил)-1- этил- 5-оксипиразола. 3 г 2-хлор-3-этилтиометил-4-метансульфонилбензойной кислоты, 0,72 г карбоната калия, 50 мл трет-амилового спирта, 1,95 г N,N"-дициклогексилкарбонимида и 4,5 г 25%-ного раствора 1-этил-5-пиразола в трет-амиловом спирте смешивали и нагревали при помешивании при 70-80оС в течение 4 ч. После охлаждения смесь дистиллировали при пониженном давлении и к остатку добавляли 200 мл воды. После отфильтрования нерастворимых компонентов фильтрат промывали хлороформом. К водному слою добавляли хлористоводородную кислоту и смесь экстрагировали хлороформом. Экстракт сушили, растворитель отгоняли и получали желаемое соединение в виде неочищенного продукта. Сырой продукт перекристаллизовывали из этанола и получали 1,88 г очищенного соединения (т.пл. 142-145оС). П р и м е р 5. Получение 4-(2-хлор-5-этансульфонилметил-4-метансульфонилбензо-ил)-1-этил-5-оксипиразо ла. 0,5 г соединения, полученного в примере 4, растворяли в растворе, содержащем 30 мл хлороформа и 30 мл тетрагидрофурана при комнатной температуре и добавляли при охлаждении в ледяной бане 2,2 эквивалента м-хлорнадбензойной кислоты. Смесь постепенно нагревали до комнатной температуры и перемешивали один день. Растворитель отгоняли и полученные кристаллы собирали фильтрацией и промывали этиловым эфиром. Получали 2,2 г целевого продукта, т.пл. 133-135оС). П р и м е р 6. Получение 4-(2-хлор-3-этансульфинил-4-метансульфонилбензоил)-1- этил -5-оксипиразола. 0,45 г соединения, полученного в примере 4, растворяли в 30 мл диоксана и добавляли 0,21 г тригидрата бромистого натрия. Смесь перемешивали при комнатной температуре 30 мин и затем добавляли воду. Смесь экстрагировали хлороформом. Экстракт сушили, растворитель отгоняли и получали неочищенный продукт. Сырой продукт очищали колоночной хроматографией (элюировали смесью хлороформ-этанол) и получали 0,2 г целевого соединения в виде маслянистого вещества. П р и м е р 7. Получение 4-(2,4-дихлор-3-метоксикарбонилбензоил)-1-этил-5-оксипи- разола. Это соединение получали таким же способом, что и в примере 2. Т.пл. 167-170оС. П р и м е р 8. Получение 5-бензилокси-4-(2,4-дихлор-3-метоксикарбонилбензоил)-1-этилпиразола. Раствор, полученный растворением 0,3 г соединения, полученного в примере 7, и 0,1 г триэтиламина в 13 мл бензола, перемешивали при комнатной температуре 30 мин и затем при 50оС 3 ч. Нерастворимые вещества отфильтровывали, фильтрат концентрировали при пониженном давлении. Концентрированный продукт очищали хроматографией на колонке силикагеля (элюировали смесью бензол-этилацетат) и получали 0,15 г желаемого соединения в виде маслянистого вещества. П р и м е р 9. Получение 5-окси-4-(4-метансульфонил-3-метоксиметил-2-метилбензо- ил) -3-метоксиметил-1-метилпиразола. 1) 5-(4-метансульфонил-3-метоксиметил-2-метилбензоил)-окси-3-метоксиметил-1- метилпиразола. 1,9 г 5-окси-3-метоксиметилпиразола добавляли к смеси, содержащей 8 мл водного раствора, содержащего 0,5 г 85%-ной гидроокиси калия и 12 мл хлороформа, и затем к ней добавляли 4-метансульфонил-3-метоксиметил-2-метилбензоилхлорид. Смесь перемешивали 3 ч при комнатной температуре. Затем реакционную смесь экстрагировали хлороформом. Хлороформовый раствор промывали водой, сушили и получали желаемое соединение довольно высокого качества в виде маслянистого вещества. 2) 5-окси-4-(4-метансульфонил-3-метоксиметил-2-метилбензоил)-3- метоксиметил-1-метилпиразола. 3,0 г соединения, полученного на стадии (1), 2,7 г карбоната калия и 8 мл диоксана перемешивали при 120оС 3,5 ч. Добавляли 20 мл воды и смесь оставляли для охлаждения. Реакционную смесь промывали хлороформом и подкисляли хлористоводородной кислотой. Реакционный раствор экстрагировали хлороформом, промывали водой, сушили и получали 1,8 г неочищенного продукта. Сырой продукт перекристаллизовывали из этанола и получали 1,2 г желаемого соединения. Т.пл. 100-104оС. П р и м е р 10. Получение 4-(3-ацетоксиметил-2-хлор-4-метансульфонилбензоил)-1-этил-5-оксипиразола. Это соединение получали таким же способом, что и в примере 2. Т.пл. 140-144оС. П р и м е р 11. Получение 4-(2-хлор-3-оксиметил-4-метансульфонил-бензоил)-1-этил- 5-оксипиразола. К 30 мл метанолового раствора, содержащего 0,3 г соединения, полученного в примере 10, добавляли 5 мл водного раствора, содержащего 0,1 г 93%-ной гидроокиси натрия и смесь перемешивали 2 ч при комнатной температуре. Метанол отгоняли при пониженном давлении. Затем добавляли хлористоводородную кислоту к полученному остатку. Выпавший в осадок продукт собирали фильтрацией и получали 0,2 г желаемого соединения. Т.пл. 70-76оС. Физические свойства соединений, полученных таким же образом, что и в предыдущих примерах, представлены в табл.3 и 4, включающих и соединения, полученные в предыдущих примерах. Номер соединения в табл.3 соответствует номеру соединения в табл.4. Если соединение согласно изобретению подлежит использованию в качестве сельскохозяйственного или садового гербицида, его обычно смешивают с подходящим носителем, например с твердым носителем, таким как глина, тальк, бентонит или диатомовая земля, или с жидким носителем, таким как вода, спирт (например, метанол или этанол), ароматический углеводород (например, бензол, толуол или ксилол), хлорированный углеводород, простой эфир, кетон, сложный эфир (например, этилацетат) или амид кислоты (например, диметилформамид). Если желают получить оптимальную композицию, такую как жидкий препарат, эмульгирующийся концентрат, смачивающийся порошок, дуст, гранула или суспензия, к соединению согласно изобретению и носителю может быть добавлен эмульгатор, диспергатор, суспендирующее средство, средство, способствующее проникновению и распределению (растеканию) композиции, или стабилизатор. Далее, если необходимо, то добавляют другие гербициды, различные инсектициды, бактерициды, регуляторы роста растений или синергические средства в композицию в процессе ее приготовления или во время применения гербицида. Другие гербициды могут быть совмещены с гербицидом согласно изобретению, например, соединения, описанные в Farm Chemicals Hondbook, the 73 rd Edition, 1987. Среди них могут быть упомянуты, например, атразин, цианазин, алахлор, метолахлор, ЕРТС, 2,4-Д, бутилат, дикамба, бромоксинил и тридифан. Далее могут быть также объединены с гербицидом согласно настоящему изобретению N-/(4,6-диметоксипиримидин-2-ил)-аминокарбонил/-3-хлор -4- метоксикарбонил-1-метилпиразол-5-суль-фонамид или N-/(4,6-диметоксипиримидин-2-ил)-аминокарбонил)-3-бром-4- метоксикарбонил-1-метилпиразол-5-сульфонамид, описанные в патенте США N 4668277. Доза варьирует в зависимости от места применения, сезона, метода применения, вида культурного растения и тому подобного. В общем доза в пределах 0,001-10 кг на 1 га активного ингредиента обычно является приемлемой. Далее следуют примеры композиций, содержащих в качестве гербицида соединения согласно изобретению, как активный ингредиент. Следует понимать, что настоящее изобретение ни в коей мере не ограничивается этими конкретными примерами. П р и м е р к о м п о з и ц и и 1. Смачивающийся порошок, мас.ч. Соединение N 3 согласно изобретению 60 Цееклит РFР (торговое название глины каолино- вого типа, производство Zuklite Industries Co, Ltd 33 Cорпол 5039 (торговое название смеси неионоген- ного и анионоактивного поверхностно-активного вещества, производимого Toho Chemical, Co Ltd 5 Карплекс (торговое название средства, предотвращающего коагуляцию, состоящего из смеси поверхностно- активного вещества и тонкодисперсного порошка кремнезема, производство Shionogi Pharmaceutical Co. Ltd 2 Перечисленные компоненты гомогенно измельчают в порошок и смешивают для получения смачивающегося порошка. П р и м е р к о м п о з и ц и и 2. Смачивающийся порошок, мас.ч. Соединение N 7 согласно изобретению 60 Цееклит РFР (торговое название глины каолинового типа, выпускаемой Zuklite Industries Co. Ltd 33 Cорпол 5039 (торговое название смеси неионогенного и анионоактивного поверхностно-активного вещества, производство Toho Chemical Cо. Ltd 5 Карплекс (торговое название средства, предотвращающего коагуляцию, образованного смесью поверхностно-активного вещества и тонкодисперсного порошка кремнезема, производимого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 3. Смачивающийся порошок, мас.ч. Соединение N 15 согласно изобретению 60 Цееклит РFР (торговое название глины каолинового типа, производимой Zuklite Industries Co. Ltd 33 Сорпол 5039 (торговое название смеси неионогенного и анионоактивного поверхностно-активного вещества, производимой Toho Chemical Co. Ltd 5 Карплекс (торговое название средства, предотвращающего коагуляцию, состоящее из смеси поверхностно-активного вещества и тонкодисперсного порошка кремнезема, производимого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 4. Смачивающийся порошок, мас.ч. Соединение согласно изобретению N 21 60 Цееклит РFР (торговое название глины каолинового типа, производство Zuklite Industries Co. Ltd 33 Cорпол 5039 (торговое название смеси неионоген- ного и анионоактивного поверхностно-активного вещества, производство Toho Chemical Co. Ltd 5 Карплекс (торговое название средства, предотвращающего коагуляцию, являющегося смесью поверхностно- активного вещества и тонко- дисперсного порошка кремне- зема, выпускаемого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 5. Смачивающийся порошок, мас.ч. Соединение N 25 согласно изобретению 60 Цееклит РFP (торговое название глины каолинового типа, выпускаемой Luklite Industries Co. Ltd 33 Cорпол 5039 (торговое название смеси неионоген- ного и анионоактивного поверхностно-активного ве- щества, выпускаемого Toho Chemical Co. Ltd 5 Карплекс (торговое название средства, предотвраща- ющего коагуляцию, состоящего из смеси поверхностно- активного вещества и тонкодисперсного порошка кремнезема, выпускаемого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 6. Смачивающийся порошок, мас.ч. Соединение N 35 согласно изобретению 60 Цееклит РFР (торговое название глины каолинового типа, выпускаемой Zuklite Industries Co. Ltd 33 Cорпол 5039 (торговое название смеси неионоген- ного и анионоактивного поверхносно-активного вещества, производимого Toho Chemical Co. Ltd 5 Карплекс (торговое название антикоагулянта, являющегося смесью поверхностно-активного вещества и тонкодисперсного порошка кремнезема, изготавливаемого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 7. Эмульгирующийся концентрат, мас.ч. Соединение N 3 согласно изобретению 1,5 Ксилол 78,6 N,N-диметилформамид 15 Сорпол 2680 (торговое название смеси неионо- генного и анионоактивного поверхностно-активного вещества, выпускаемого Togo Chemical Co. Ltd 5 Перечисленные выше ингредиенты гомогенно смешивали для получения эмульгирующего концентрата. П р и м е р к о м п о з и ц и и 8. Эмульгирующийся концентрат, мас.ч. Соединение N 11 согласно изобретению 1,5 Ксилол 78,5 N,N-диметилформамид 15 Сорпол 2680 (торговое название смеси неионогенного и анионо- активного поверхностно- активного вещества, производимого 5 П р и м е р к о м п о з и ц и и 9. Суспензия, мас.ч. Соединение N 3 согласно изобретению 40 Агризол В-710 (торговое название неионогенного поверхностно-активного вещества, выпускаемого Kao Corporation 10 Рунокс 1000 С (торговое название анионоактив- ного поверхностно- активного вещества, выпуска- емого Toho Chemical Co. Ltd 0,5 1% Родопол-вода (торговое название загустителя, выпускаемого Rhone-Paulenc 20 Вода 29,5 Перечисленные ингредиенты гомогенно смешивали для получения суспензии. П р и м е р к о м п о з и ц и и 10. Суспензия, мас.ч. Соединение N 10 согласно изобретению 40 Агризол В-710 (торговое название неионогенного поверхностно-активного вещества, выпускаемого Као Cоrporation 10 Рунокс 1000 С (торговое название неионоактивного поверхностно-активного вещества, выпускаемого Toho Chemical Co. Ltd 0,5 1% Родопол-вода (торговое название загустителя, выпускаемого Rhone Poulenс 20 Вода 29,5 П р и м е р к о м п о з и ц и и 11. Жидкая композиция, мас.ч. Соединение N 39 согласно изобретению 30 Ниппол (торговое название неионогенного поверхностно-активного вещества, выпускаемого Nissan Chemical Industries, Ltd 10 Вода 60 Перечисленные выше компоненты гомогенно смешивали для получения жидкой композиции. П р и м е р к о м п о з и ц и и 12. Жидкая композиция, мас.ч. Соединение N 40 согласно изобретению 30 Ниппол (торговое название неионогенного поверхностно-активного вещества, производимого Nissan Chemical Indusctries, Ltd 10 Вода 60 П р и м е р к о м п о з и ц и и 13. Жидкая форма, мас.ч. Соединение N 46 согласно изобретению 30 Ниппол (торговое название неионогенного поверхностно-активного вещества, выпускаемого Nissan Chemical Industries, Ltd 10 Вода 60 П р и м е р к о м п о з и ц и и 14. Жидкая форма, мас.ч. Соединение N 41 согласно изобретению 10 Сорпол W-150 (торговое название неионогенно- го поверхностно-активного вещества, производимого Toho Chemical Co. Ltd 10 Вода 80 Перечисленные выше ингредиенты гомогенно смешивали для получения жидкой формы препарата. При использовании упомянутые выше смачивающиеся порошки, эмульгирующиеся концентраты, суспензии и жидкие формы композиций разбавляют водой в 50-1000 раз и применяют в количествах, обеспечивающих 0,001-5 кг активного ингредиента на 1 га. Соединения согласно изобретению применимы не только на сельскохозяйственных и садовых плантациях, таких как суходольные поля, рисовые поля и фруктовые сады, но и на площадях, изъятых из сельскохозяйственного употребления, таких как стадионы, пустоши и полосы отчуждения вдоль железных дорог для борьбы с различными сорняками. Доза при их применении варьирует в зависимости от применяемого метода, обрабатываемой площади, сезона применения, вида культурных растений и так далее. Однако обычно она находится в пределах 0,001-5,0 кг на 1 га. Далее гербицидные активности соединений согласно изобретению будут описаны со ссылкой на конкретные примеры испытаний. П р и м е р и с п ы т а н и й 1. Тест на гербицидное действие при обработке почвы. Пластиковый ящик длиной 22 см, шириной 15 см и глубиной 6 см заполняли дилювиальной почвой, подвергающейся стерилизации, и в нее высевали семена Echinochloa crus-galli, Setaria viridis, Eleusine indica, Digitaria adscendens, Panicum dichotomiflorum, Abutilon theophrasti, Amaranthus lividus, Polygonum longisetum и Zea maus и высаживали клубни Сyperus esculentus. Семена засыпали слоем почвы толщиной около 1,5 см и затем на поверхность почвы равномерно наносили раствор гербицида, обеспечивающий заданную концентрацию активного ингредиента. Гербицидный раствор готовили разбавлением водой смачивающегося порошка, эмульгирующегося концентрата, жидкой композиции или суспензии и наносили с помощью ручного распылителя на всю поверхность почвы. Через три недели после применения гербицидного раствора определяли гербицидное действие против каждого вида сорняков на основе следующих стандартных оценок: полученные результаты представлены в табл.5. Номер соединения в табл.5 соответствует номеру соединения в табл.3. Стандартные оценки: 5 степень подавления роста более 90% (почти полное увядание) 4 степень подавления роста 70-90% 3 степень подавления роста 40-70% 2 степень подавления роста 20-40% 1 степень подавления роста 5-20% 0 степень подавления роста менее 5% (почти неэффективно). Упомянутые оценки подавления роста рассчитывали по следующему уравнению: степень подавления роста (%) (1 Т/N) х 100, где Т вес зеленой массы над поверхностью почвы обработанного участка; N зеленой массы над поверхностью почвы необработанного участка. П р и м е р и с п ы т а н и й 2. Тест на гербицидное действие при обработке листьев. Пластиковые ящики длиной 22 см, шириной 15 см и глубиной 6 см заполняли стерилизованной дилювиальной почвой и семена Echinochloa crus-galli, Setaria viridis, Eleusine indica, Digitaria adscendens, Panicum dichotomiflorum, Xanthium strumarium, abutilon theophrasti, Amaranthus lividus, Polygonum longisetum и Zea mays высевали местами и высаживали клубни Сyperus esculentus. Затем семена покрывали слоем почвы толщиной около 1,5 см. Сорняки и культурные растения выращивали до стадии 2 или 3 листа и гербицидный раствор равномерно разбрызгивали на листья так, чтобы активный ингредиент был применен в заранее заданной концентрации. Гербицидный раствор готовили разбавлением смачивающегося порошка, эмульгирующегося концентрата, жидкой формы композиции или суспензии, описанных в примерах получения композиций, водой и наносили на всю поверхность листьев сорняков и культурных растений с помощью небольшого распылителя. Через две недели после применения гербицидного раствора определяли гербицидное действие против каждого вида сорняков на основе стандартных оценок, описанных в примере испытаний 1, и фитотоксичность против каждого вида культурных растений определяли также на основе стандартных оценок, приведенных в примере испытаний 1. Результаты представлены в табл.6. Номер соединений в табл. 6 соответствует номеру соединения в табл.3. В табл. 5 и 6 применены следующие сокращения: доза доза активного ингредиента (г/ар) ЕС Echinochloa crus-galli (просо куриное) SE Setaria viridis (щетинник зеленый) EL Eleusine indica (элеузина индийская) DI Digitaria adscendens (росичка кровяная) РА Panicum dichotomiflorum (просо) АВ Abutilon theophrasti (канатник Теофраста) АМ Amaranthus lividus (ширица синеватая) PO Polygonum longisetum (горец длинностолбиковый) ХА Kanthum strumarium (дурнишник) СУ Сyperus esculentus (сыть съедобная) ZE Zea mays (кукуруза) 2,47 (3Н, с), 3,22 (3Н,с), 3,50 (3Н,с), 3,69 (3Н,с), 4,96 (2Н,с), 7,30 (1Н,с), 7,78 (2Н, А-В кв), 10,9 (1Н) /СDCl3/ 2,41 (3Н,с), 3,17 (3Н,с), 3,50 (3Н,с), 4,94 (2Н,с), 5,53 (2Н,с), 7,30-8,12 (8Н,м) /CDCl3) 1,44 (3Н, т), 2,48 (3Н,с), 3,23 (3Н,с), 3,51 (3Н,с), 4,07 (2Н,кв), 4,98 (2Н,с), 7,36 (1Н,с), 7,82 (2Н, А-В кв) /CDCl3/ 1,48 (6Н, д), 2,47 (3Н,с), 3,19 (3Н,с), 3,48 (3Н,с), 4,53 (1Н,м), 4,92 (2Н,с), 7,18 (1Н,с), 7,69 (2Н, А-В кв), 9,57 (1Н) /CDCl3/ 2,43 (3Н,с), 3,78 (3Н,с), 3,94 (3Н,с), 7,24-7,81 (7Н,м), /CDCl3/ 1,07 (3Н, т), 2,27 (3Н,с), 3,01 (3Н,с), 3,30-3,65 (5Н,м), 4,77 (2Н,с), 6,99 (1Н,с), 7,48 (А-В кв), 8,22 (1Н,с) /CDCl3/ 1,45 (3Н,т), 3,04 (2Н,кв), 4,05 (2Н,кв), 4,91 (2Н,кв), 7,29 (1Н,с), 7,70 (1Н,с), 7,85 (2Н,кв) (CDCl3) 0,91-2,03 (8Н,м), 2,41 (3Н,с), 3,20 (3Н,с), 3,91-4,47 (4Н,м), 7,36-8,08 (4Н,м), (CDCl3). 1,26 (3Н, т), 1,49 (3Н,д), 2,49 (3Н,с), 3,24 (3Н,с) 3,69 (2Н,кв), 4,59 (1Н,м), 5,00 (2Н,с), 7,28-8,14 (4Н,м), /CDCl3/ 1,27 (3Н, т), 1,67 (3Н,с), 2,42 (3Н,с), 3,23 (3Н,с), 3,63 (3Н,с), 3,68 (2Н,кв), 5,02 (2Н,с), 7,7 (1Н,с), 7,75 (2Н, А-В кв), (СDCl3) 1,45 (3Н,т), 2,49 (шир.с), 3,44 (3Н,с), 4,03 (2Н,кв), 4,66 (2Н,с), 7,29 (2Н,кв.), 7,39 (1Н,с); 1,47 (3Н, т), 3,29 (3Н,с), 3,51 (3Н,с),3,82 (3Н,с), 4,09 (2Н,кв), 5,00 (2Н,с), 7,50 (1Н,с), 7,84 (2Н, А-В кв), 7,96 (1Н, с), /CDCl3/ 1,07-1,67 (9Н, м), 2,59 (3Н,с), 3,17 (3Н,с), 3,42 (2Н,т), 3,96 (2Н,т), 5,61 (1Н,кв), 7,27 (1Н,с), 7,61 (2Н,А-В кв), 9,66 (1Н, широкий с) /CDCl3/ 1,09 (3Н, т), 1,45 (3Н,т), 2,55 (3Н,с), 3,16 (3Н,с) 3,27 (3Н,с), 3,70-4,20 (4Н, м), 5,13-5,36 (1Н, м) 7,14 (1Н,с), 7,60 (2Н, А-В кв), 9,46 (1Н, широкий с) /СDCl3/ Далее получение бензойных кислот будет описано со ссылкой на ссылочные примеры. Однако следует понимать, что изобретение ни в коей мере не ограничено такими конкретными примерами. С с ы л о ч н ы й п р и м е р 1. Получение 4-метансульфонил-3-метоксиметил-2-метил- бензойной кислоты и 3-метоксиметил-2-метил-4-метилтиобензойной кислоты. 1) 2-метил-3-нитробензиловый спирт. 39,0 г (0,2 моля) метилового эфира 2-метил-3-нитробензойной кислоты растворяли в 600 мл трет-бутанола и к ним добавляли 19,0 г боргидрида натрия. В реакционную смесь при нагревании с обратным холодильником по каплям добавляли 150 мл метанола в течение 1 ч. Нагревание с обратным холодильником продолжали еще один час для завершения реакции. Реакционную смесь оставляли для охлаждения и затем в нее добавляли воду. Растворитель отгоняли при пониженном давлении. К остатку добавляли воду и хлороформ, органический слой отделяли и сушили над безводным сульфатом натрия. Затем отгоняли растворитель и получали 30,7 г 2-метил-3-нитробензилового спирта. 2) 2-метил-3-нитробензилметиловый эфир. 30,1 г (0,18 моля) 2-метил-3-нитробензилового спирта, полученного на предыдущей стадии, растворяли в 200 мл бензола и в раствор последовательно добавляли 0,2 г бромистого тетра-н-бутиламмония и 20,1 г 50%-ного водного раствора гидроокиси натрия. Затем при комнатной температуре по каплям добавляли 27,2 г диметилсульфата. Дальше реакцию проводили 3 ч при перемешивании. К реакционному раствору добавляли воду и органический слой отделяли, последовательно промывали водой, 2%-ным водным раствором хлористоводородной кислоты, водой и насыщенным водным раствором хлористого натрия. Затем растворитель отгоняли и получали 30,9 г 2-метил-3-нитробензилметилового эфира в виде маслянистого вещества. 3) 3-метоксиметил-2-метиланилин. К 30,7 г (0,17 моля) полученного выше 2-метил-3-нитробензилметилового эфира добавляли 200 мл метанола. После растворения соединения в метаноле в него постепенно добавляли 92 мл концентрированной хлористоводородной кислоты. Затем в реакционную смесь постепенно добавляли 30,4 г железного порошка так, чтобы температура реакции не превышала 60оС и реакцию продолжали еще 1 ч. К реакционному раствору добавляли 300 мл воды и добавляли гидроокись натрия до тех пор, пока рН не станет более 8. К полученному шламму добавляли хлороформ и смесь тщательно перемешивали. Затем твердое вещество отделяли фильтрацией и органический слой отделяли от фильтрата. Органический слой последовательно промывали водой и насыщенным водным раствором хлористого натрия и затем сушили над безводным сульфатом натрия. Далее растворитель отгоняли при пониженном давлении и получали 23,1 г 3-метоксиметил-2-анилина в виде маслянистого вещества. 4) 3-метоксиметил-2-метил-4-тиоцианоанилин. 22,6 г (0,15 моля) 3-метоксиметил-2-метиланилина растворяли в 300 мл метанола. Затем в раствор добавляли 36,5 г тиоцианата натрия для получения однородного раствора. Этот раствор охлаждали до 0оС и в него по каплям добавляли 100 мл насыщенного метанольного раствора бромистого натрия с 25,2 г брома так, чтобы температура реакции не превышала 5оС. После капельной добавки смесь перемешивали при температуре, не превышающей 5оС 1 ч и при комнатной температуре один час для завершения реакции. Реакционный раствор выливали в 1 л воды и нейтрализовали 5%-ным водным раствором карбоната натрия. Добавляли хлороформ для экстрагирования маслянистого вещества. Хлороформовый слой промывали водой и насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом натрия. Затем растворитель отгоняли при пониженном давлении и получали 29,6 г желаемого продукта. 5) 3-метоксиметил-2-метил-4-метилтиоанилин. 29,1 г (0,14 моля) 3-метоксиметил-2-метил-4-тиоцианоанилина растворяли в 200 мл этанола и смешивали со 100 мл водного раствора, содержащего 33,6 г нонагидрата сульфата натрия при комнатной температуре. Затем в полученный раствор по каплям добавляли 21,9 г иодистого метила и смесь оставляли на 3 ч при комнатной температуре для продолжения реакции. После завершения реакции растворитель отгоняли при пониженном давлении и к остатку добавляли воду и хлороформ. Затем отделяли органический слой и последовательно промывали водой и насыщенным водным раствором хлористого натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и получали 25,6 г желаемого продукта в виде маслянистого вещества. 6) 3"-иодо-2"-метил-6"-диметилтиобензилметиловый эфир. К 25,6 г (0,13 моля) 3-метоксиметил-2-метил-4-метилтиоанилина добавляли 100 мл воды и 33 мл концентрированной хлористоводородной кислоты для превращения его в гидрохлорид анилина. Этот раствор охлаждали до 0оС и в него по каплям добавляли 30 мл водного раствора, содержащего 9,3 г нитрита натрия так, чтобы температура реакции не превышала 5оС. После завершения капельной добавки перемешивание продолжали еще 30 мин для завершения диазотирования. 100 мл водного раствора, содержащего 33 г иодистого калия, нагревали до 70оС и водный раствор соли диазония, полученного выше, постепенно добавляли к нему и разлагали. Реакционный раствор далее перемешивали 1 ч при 70оС и затем оставляли для охлаждения. Масляный компонент экстрагировали бензолом. Бензольный слой промывали последовательно водой, насыщенным раствором кислого сульфита натрия, водой и насыщенным раствором хлористого натрия в воде. Затем растворитель отгоняли при пониженном давлении и остаток очищали колоночной хроматографией (элюентом служил бензол). Получали 30,0 г желаемого продукта, точка плавления (т.пл.) 56-59оС. 7) 3-метоксиметил-2-метил-4-метилтиобензойная кислота. 27,7 г (0,09 моля) 3"-иодо-2"-метил-6"-метилтиобензилметилового эфира растворяли в 100 мл осушенного тетрагидрофурана и к раствору по каплям добавляли при -70оС 63 мл 1,5 М н-бутиллития в н-гексане. По окончании капельной добавки смесь перемешивали 15 мин при той же температуре и через реакционный раствор осторожно продували газообразную двуокись углерода до прекращения выделения тепла реакционным раствором. По окончании реакции температуру раствора доводили до комнатной температуры и добавляли воду и диэтиловый эфир для жидкостного разделения. Полученный водный слой промывали далее дважды диэтиловым эфиром, а затем концентрированной хлористоводородной кислотой устанавливали рН 1. Выпавшие в осадок кристаллы собирали фильтрацией, тщательно промывали водой, сушили и получали 14,4 г желаемого продукта. Т.пл. 192,0-194,0оС. 8) 4-метансульфонил-3-метоксиметил-2-метилбензойная кислота. К 11,3 г (0,05 моля) 3-метоксиметил-2-метил-4-метилтиобензойной кислоты добавляли 120 мл уксусной кислоты и 120 мл 35%-ного водного раствора перекиси водорода и смесь подвергали реакции при 80оС в течение 1 ч. После охлаждения реакционный раствор выливали в воду со льдом, после чего выпавшие в осадок кристаллы собирали фильтрацией, промывали водой, сушили и получали 12,3 г целевого соединения. Т.пл. 129,0-131,0оС. С с ы л о ч н ы й п р и м е р 2. Получение 3-метоксикарбонил-2-метил-4-метилтиобен- зойной кислоты и 4-метансульфонил-3-метоксикарбонил-2-метил-бензойной кислоты. 1) Метиловый эфир 3-амино-2-метилбензойной кислоты. 40 г метилового эфира 2-метил-3-нитробензойной кислоты растворяли в 120 мл метанола и в раствор добавляли 157 г концентрированной хлористоводородной кислоты. Затем добавляли постепенно 36,8 г железного порошка, поддерживая температуру не выше 60оС. Смесь перемешивали при комнатной температуре 4 ч и затем выливали в 1 л воды с льдом. Раствор нейтрализовали карбонатом натрия и экстрагировали хлороформом (после отфильтровывания нерастворимой фракции). Экстракт промывали насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 27,8 г желаемого соединения в виде маслянистого вещества. 2) Метиловый эфир 3-амино-2-метил-6-тиоцианобензойной кислоты. К раствору, содержащему 27,7 г метилового эфира 3-амино-2-метилбензойной кислоты, 41,5 г тиоцианата натрия и 250 мл метанола при температуре не выше 0оС по каплям медленно добавляли 100 мл насыщенного бромистым натрием метанола с 28,1 г бpома. Смесь перемешивали при комнатной температуре три часа и затем выливали в 1 л воды с льдом. Раствор нейтрализовали карбонатом натрия и экстрагировали хлороформом. Экстракт промывали насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 34,0 г желаемого продукта в виде маслянистого вещества. 3) Метиловый эфир 3-амино-2-метил-6-метилтиобензойной кислоты. К раствору, содержащему 39,5 г нонагидрата сульфида натрия и 110 мл воды по каплям добавляли раствор, содержащий 32,9 г метилового эфира 3-амино-2-метил-6-тиоцианобензойной кислоты и 300 мл этанола. Смесь перемешивали при комнатной температуре 1,5 ч и к нему по каплям добавляли 24,0 г иодистого метила при охлаждении льдом. Смесь перемешивали далее при комнатной температуре еще 2 ч и затем концентрировали при пониженном давлении. К ней добавляли насыщенный водный раствор хлористого натрия и смесь экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Затем отгоняли растворитель и получали 30,1 г желаемого соединения в виде маслянистого вещества. 4) Метиловый эфир 3-иодо-2-метил-6-метилтиобензойной кислоты. 28 г метилового эфира 3-амино-2-метил-6-метилтиобензойной кислоты перемешивали в 150 мл концентрированной хлористоводородной кислоты при комнатной температуре в течение 2 ч, чтобы превратить ее в гидрохлорид. Затем, поддерживая температуру смеси не выше 0оС, к ней по каплям добавляли раствор, содержащий 11,9 г нитрита натрия и 20 мл воды, чтобы получить раствор соли диазония. Раствор соли диазония по каплям добавляли к раствору, содержащему 28,4 г иодистого калия и 90 мл воды, поддерживая температуру раствора 80оС. По окончании капельного введения смесь перемешивали при 80оС в течение 15 мин и оставляли для охлаждения. В нее добавляли воду и смесь экстрагировали хлороформом. Экстракт промывали водным раствором кислого сульфита натрия и водой, затем сушили над безводным сульфатом натрия. Растворитель отгоняли и получали 40 г желаемого соединения в виде сырого продукта. Сырой продукт подвергали очистке колоночной хроматографией на силикагеле (элюируя бензолом) и получали 36,0 г очищенного соединения в виде маслянистого вещества. 5) 3-метоксикарбонил-2-метил-4-метилтиобензойная кислота. Поддерживая температуру раствора, содержащего 20,0 г метилового эфира 3-иодо-2-метил-6-метилтиобензойной кислоты и 70 мл осушенного тетрагидрофурана, не выше -60оС в атмосфере азота, к нему по каплям добавляли 42 мл 1,5 М раствора н-бутиллития в н-гексане. Через 15 мин в смесь вдували осушенный углекислый газ, поддерживая температуру не выше -50оС. После продувки азотом с целью очистки от газообразной двуокиси углерода в смесь по каплям добавляли 12,7 г диизопропиламина и смесь перемешивали до достижения комнатной температуры. Смесь концентрировали при пониженном давлении. В нее добавляли воду и промывали смесь хлороформом. Водный раствор подкисляли концентрированной хлористоводородной кислотой и затем экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Растворитель отгоняли и получали 7,5 г желаемого соединения. Т.пл. 178-178,5оС. 6) 4-метансульфонил-3-метоксикарбонил-2-метилбензойная кислота. Раствор, содержащий 5,0 г 3-метоксикарбонил-2-метил-4-метилтиобензойной кислоты, 25 мл уксусной кислоты и 25 мл 35%-ной перекиси водорода, перемешивали при 80оС 3 ч. После охлаждения смесь выливали в воду с льдом и экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 5,1 г желаемого соединения. Т.пл. 151-152оС. С с ы л о ч н ы й п р и м е р 3. Получение 2-хлор-3-этилтиометил-4-метансульфонил-бензойной кислоты. 1) Метиловый эфир 3-бромметил-2-хлор-4-метансульфонилбензойной кислоты. 12,1 г метилового эфира 2-хлор-4-метансульфонил-3-метилбензойной кислоты растворяли в 250 мл четыреххлористого углерода и раствор нагревали с обратным холодильником при перемешивании. Затем в него постепенно добавляли 7,5 г брома и 1,0 г перекиси бензоила в течение 30 мин и далее раствор нагревали с обратным холодильником 4 ч. После охлаждения в него добавляли 200 мл хлороформа и смесь промывали 5%-ным водным раствором кислого сульфита натрия. Органический слой отделяли и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и получали неочищенный продукт. Неочищенный продукт промывали этиловым эфиром и получали 13,2 г кристаллов желаемого соединения. Т.пл. 77-78оС. 2) Метиловый эфир 2-хлор-3-этилтиометил-4-метансульфонилбензойной кислоты. К 100 мл тетрагидрофурана добавляли 1,3 г этантиола и 1,5 г карбоната калия и затем 4,4 г метилового эфира 3-бромметил-2-хлор-4-метансульфонилбензойной кислоты и смесь перемешивали один день при комнатной температуре. Затем смесь перемешивали еще 1 ч при температуре 50-60оС. После охлаждения в нее добавляли хлороформ и смесь промывали разбавленным водным раствором карбоната калия. Хлороформовый слой отделяли и сушили. Затем растворитель отгоняли и получали 4,1 г метилового эфира 2-хлор-3-этилтиометил-4-метансульфонилбензойной кислоты в виде маслянистого вещества. 3) 2-хлор-3-этилтиометил-4-метансульфонилбензойная кислота. К раствору смеси, содержащему 50 мл 10%-ного водного раствора гидроокиси натрия и 150 мл метанола, добавляли 3,9 г метилового эфира 2-хлор-3-этилтиометил-4-метансульфонилбензойной кислоты и смесь перемешивали при комнатной температуре 30 мин. Метанол отгоняли при пониженном давлении и к остатку добавляли разбавленную хлористоводородную кислоту для преципитации кислоты. Смесь экстрагировали этилацетатом и экстракт сушили. Затем растворитель отгоняли и получали 3,5 г желаемого соединения. Т.пл. 172-174оС. С с ы л о ч н ы й п р и м е р 4. Получение 2-хлор-4-метансульфонил-3-метоксиметил- бензойной кислоты. 1) Метиловый эфир 2-хлор-4-метансульфонил-3-метоксиметилбензойной кислоты. К раствору, содержащему 12,0 г метилового эфира 3-бромметил-2-хлор-4-метан-сульфонилбензойной кислоты, полученному по ссылочному примеру 3 (1), и 100 мл метанола добавляли 50 мл метанолового раствора, содержащего 1,7 г метилата натрия и смесь перемешивали при комнатной температуре, оставляя на ночь. Растворитель отгоняли при пониженном давлении. Затем добавляли к остатку разбавленную хлористоводородную кислоту и смесь экстрагировали хлороформом. Экстракт промывали водой и сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 9,5 г неочищенного продукта. Неочищенное желаемое соединение очищали хроматографией на колонке силикагеля (элюировали бензолом) и получали 7,5 г очищенного желаемого соединения в виде маслянистого вещества. 2) 2-хлор-4-метансульфонил-3-метоксиметилбензойная кислота. К раствору, содержащему 3,0 г метилового эфира 2-хлор-4-метансульфонил-3-метоксиметилбензольной кислоты и 20 м метанола, добавляли раствор, содержащий 0,57 г 93%-ной гидроокиси натрия и 2 мл воды и смесь перемешивали при комнатной температуре 30 мин. После добавления 10 мл воды смесь концентрировали при пониженном давлении. Затем в нее добавляли разбавленной хлористоводородной кислоты и смесь экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Затем растворитель отгоняли и получали 2,6 г целевого соединения. Т.пл. 137-141оС. С с ы л о ч н ы й п р и м е р 5. Получение 2-хлор-4-метансульфонил-3-метоксиметил- бензойной кислоты (альтернативный метод способу, описанному в ссылочном примере 4). Желаемое соединение получали таким же образом, что и в ссылочном примере 1. Т.пл. 137-141оС. Физические свойства промежуточных соединений следующие: 2-хлор-3-нитробензиловый спирт: масляное вещество; 2"-хлор-3"-нитробензилметиловый эфир: маслянистое вещество; 2-хлор-3-метоксиметиланилин: маслянистое вещество; 2-хлор-3-метоксиметил-4-тиоцианоани- лин: т.пл. 90-96оС; 2-хлор-3-метоксиметил-4-метилтиоани- лин: маслянистое вещество; 2"-хлор-3"-иодо-6"-метилтиобензилме-тиловый эфир, т.пл. 53-56оС. С с ы л о ч н ы й п р и м е р 6. Получение 2-хлор-4-метансульфонил-3-метоксикарбо- нилбензойной кислоты. Желаемое соединение получали таким же образом, что и в ссылочном примере 2. Т.пл. 160-162оС. Физические свойства промежуточных соединений следующие: метиловый эфир 3-амино-2-хлорбензойной кислоты, маслянистое вещество; метиловый эфир 3-амино-2-хлор-6-тиоцианобензойной кислоты, т.пл. 80-83оС; метиловый эфир 3-амино-2-хлор-6-метилтиобензойной кислоты, т.пл. 70-72оС; метиловый эфир 2-хлор-3-иод-6-метилтиобензойной кислоты, маслянистое вещество; 2-хлор-3-метоксикарбонил-4-метилтио- бензойная кислота, т.пл. 176-179оС. С с ы л о ч н ы й п р и м е р 7. Получение 4-метансульфонил-3-/(2-метоксиэтил)-окси- карбонил/-2-метилбензойной кислоты. Желаемое соединение было получено таким же образом, что и в ссылочном примере 2. Т.пл. 118-121оС. Физические свойства промежуточных соединений следующие: 2-метоксиэтиловый эфир 3-амино-2-метилбензойной кислоты, маслянистое вещество; 2-метоксиэтиловый эфир 3-амино-2-метил-6-тиоцианобензойной кислоты, т. пл.79-81оС; 2-метоксиэтиловый эфир 3-амино-2-метил-6-метилтиобензойной кислоты, маслянистое вещество; 2-метоксиэтиловый эфир 3-иодо-2-метил-6-метилтиобензойной кислоты, маслянистое вещество; 3-/(2-метоксиэтил)оксикарбонил/-2-ме- ти-4-метилтиобензойзная кислота, т.пл. 90-93оС. С с ы л о ч н ы й п р и м е р 8. Получение 2-метил-4-метилтио-З-н-пропоксикарбонилбен- зойной кислоты и 4-метансульфонил-2-метил-3-н-пропоксикарбонилбензойной кислоты 1) Метиловый эфир 3-бром-2-метил-6-метилтиобензойной кислоты. 16,1 г соединения, полученного в ссылочном примере 2 (3), перемешивали в 150 мл бромистоводородной кислоты (48%) для превращения его в бромгидрат. Поддерживая температуру не выше, чем 0оС, к полученному раствору по каплям добавляли раствор, содержащий 7,2 г нитрита натрия и 20 мл воды для получения раствора соли диазония. Раствор соли диазония по каплям добавляли к раствору, содержащему 6,0 г бромида меди и 7,7 г 48%-ной бромистоводородной кислоты, нагревая с обратным холодильником. После завершения капельной добавки смесь нагревали с обратным холодильником еще 1 ч и оставляли для охлаждения. В нее добавляли воду с льдом и смесь экстрагировали хлороформом. Экстракт промывали водным раствором кислого сульфита натрия и водой и затем сушили над безводным сульфатом натрия. Растворитель отгоняли и получали 19,2 г желаемого соединения в виде неочищенного продукта. Неочищенный продукт очищали хроматографией на колонке силикагеля (проявляли бензолом) и получали 17,1 г чистого продукта в виде маслянистого вещества. 2) 3-бром-2-метил-6-метилтиобензойная кислота. К 100 мл этанолового раствора, содержащего 17,0 г метилового эфира 3-бром-2-метил-6-метилтиобензойной кислоты, добавляли 16 г 50%-ного водного раствора гидроокиси натрия и смесь нагревали с обратным холодильником 3 ч. Реакционную смесь концентрировали при пониженном давлении. Затем в нее добавляли воду и смесь промывали хлороформом. Водный слой подкисляли концентрированной хлористоводородной кислотой и экстрагировали хлороформом. Экстракт сушили над безводным сульфатом натрия. Растворитель отгоняли и получали 15,9 г желаемого соединения. Т.пл. 98-103оС. 3) н-пропиловый эфир 3-бром-2-метил-6-метилтиобензойной кислоты. Хлористый тионил добавляли к 15,8 г 3-бром-2-метил-6-метилтиобензойной кислоты и смесь нагревали с обратным холодильником 4 ч. Хлористый тионил отгоняли и к остатку добавляли 70 мл н-пропанола при охлаждении льдом. Затем добавляли по каплям раствор, содержащий 7,3 г пиридина и 20 мл н-пропанола. Смесь оставляли на ночь при перемешивании при комнатной температуре и концентрировали при пониженном давлении. Затем добавляли этилацетат и смесь последовательно промывали 5% -ным водным раствором карбоната натрия, 10%-ной хлористоводородной кислотой и водой и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и получали 18,0 г желаемого соединения в виде неочищенного продукта. Сырой продукт очищали хроматографией на колонке силикагеля (элюировали бензолом) и получали 16,6 г очищенного продукта в виде маслянистого вещества. 4) 3-бром-2-метил-6-метилтиобензойная кислота. Это соединение получали таким же образом, что и в ссылочном примере 2 (5). Т.пл. 138-142оС. 5) 3-бром-6-метансульфонил-2-метилбензойная кислота. Это соединение получали таким же образом, что и в ссылочном примере 2 (6). Т.пл. 142-146оС. С с ы л о ч н ы й п р и м е р 9. Получение 2-хлор-3-изопропоксикарбонил-4-метансуль- фонилбензойной кислоты. Это соединение получали из соединения по ссылочному примеру 6 (4) таким же образом, что и в ссылочном примере 8 (2). Т.пл. 146-148оС. Физические свойства промежуточных соединений следующие: 2-хлор-3-иодо-6-метилтиобензойная кислота, т.пл. 155-159оС; изопропиловый эфир 2-хлор-3-иодо-6-метилтиобензойной кислоты, маслянистое вещество; 2-хлор-3-изопропоксикарбонил-4-ме- тилтиобензойная кислота, т.пл. 114-118оС. С с ы л о ч н ы й п р и м е р 10. Получение 3-(1-метоксиэтил)-2-метил-4-метилтиобен-зойной кислоты и 4-метансульфонил-3-(1-метоксиэтил)-2-метилбензойной кислоты. 1) 2"-метил-3"-нитроацетофенон. К 5,4 г металлического магния добавляли по каплям 5 мл абсолютного этанола и 0,5 мл четыреххлористого углерода в потоке сухого азота. Далее добавляли 130 мл сухого диэтилового эфира при нагревании с обратным холодильником и затем по каплям добавляли раствор, содержащий 25 мл диэтилового эфира, 35,2 г диэтилового эфира малоновой кислоты и 20 мл этанола, со скоростью, поддерживающей температуру образования флегмы. После завершения капельного введения нагревание с обратным холодильником продолжали 3 ч для получения диэтилового эфира этоксимагнезиомалоновой кислоты. К полученному таким образом раствоpу диэтилового эфира этоксимагнезиомалоновой кислоты по каплям добавляли 150 мл раствора диэтилового эфира, содержащего 40 г хлорида 2-метил-3-нитробензойной кислоты, полученного из 2-метил-3-нитробензойной кислоты и хлористого тионила, в течение 20 мин, при нагревании с обратным холодильником и реакцию продолжали 2 ч. После охлаждения добавляли разбавленную серную кислоту для проведения гидролиза. Слой диэтилового эфира последовательно промывали водой и насыщенным водным раствором хлористого натрия. Затем растворитель отгоняли при пониженном давлении, остаток сушили и получали неочищенное соединение диэтиловый эфир 2-(2-метил-3-нитробензоил)-малоновой кислоты. В этот неочищенный продукт добавляли смесь, содержащую 7,5 мл концентрированной серной кислоты, 60 мл уксусной кислоты и 40 мл воды и смесь нагревали с обратным холодильником 6 ч. Затем рН смеси устанавливали равным 10 с помощью 20%-ного раствора гидроокиси натрия в воде. Выпавший в осадок масляный компонент экстрагировали хлороформом. Хлороформовый слой последовательно промывали водой и водным раствором хлористого натрия. Затем растворитель отгоняли при пониженном давлении и получали 34,0 г желаемого продукта. (Выход 95%), т.пл. 53-54оС. 2) Получение 1-метил-2-метил-3-нитробензилового спирта. К 50 мл метанолового раствора 0,5 г гидроокиси натрия добавляли при 0оС 0,9 г боргидрида натрия и затем к нему по каплям добавляли 100 мл метанолового раствора 14,3 г 2-метил-3-нитроацетофенона. Температуру смеси повышали до комнатной температуры и реакцию продолжали 1 ч. По завершении реакции реакционную смесь выливали в воду и экстрагировали бензолом. Последующую операцию проводили обычным образом и получали 14,3 г желаемого продукта в виде маслянистого вещества (выход 99%). В дальнейшем проводили синтез таким же образом, что и в ссылочном примере 1, для получения промежуточных соединений (3) (9). 3) 1-метил-2"-метил-3"-нитробензилметиловый эфир, маслянистое вещество. 4) 1-метил-3"-амино-2"-метилбензилметиловый эфир, маслянистое вещество. 5) 1-метил-3-амино-2-метил-6-тиоцианобензилметиловый эфир, твердое вещество. 6) 1-метил-3"-амино-2"-метил-6"-метилтиобензилметиловый эфир, маслянистое вещество. 7) 1-метил-3"-иодо-2"-метил-6"-метилтиобензил, маслянистое вещество. 8) 3-(1-метоксиэтил)-2-метил-4-метилтиобензойная кислота, маслянистое вещество, 9) 4-метансульфонил-3-(1-метоксиэтил)-2-метилбензойная кислота, т.пл. 106-109оС. С с ы л о ч н ы й п р и м е р 11. Получение 2,4-дихлор-3-метоксикарбонилбензойной кислоты. 1) 2,4-дихлор-3-нитробензойная кислота. К раствору 25 мл дымящей азотной кислоты и 20 мл серной кислоты постепенно добавляли 25 г 2,4-дихлорбензойной кислоты. После прекращения выделения тепла реакционную смесь выливали в воду с льдом. Твердый осадок промывали водой, сушили и получали 23,0 г желаемого соединения. 2) Метиловый эфир 2,4-дихлор-3-нитробензойной кислоты. 23,0 г 2,4-дихлор-3-нитробензойной кислоты и 150 мл хлористого тионила нагревали с обратным холодильником 6 ч. Затем хлористый тионил отгоняли и получали неочищенный 2,4-дихлор-3-нитробензоилхлорид. К неочищенному соединению добавляли 200 мл метанола и нагревали с обратным холодильником. Метанол отгоняли и добавляли этилацетат для получения этилацетатного раствора. Раствор последовательно промывали 5%-ным водным раствором гидроокиси натрия, разбавленной хлористоводородной кислотой и водой. После осушения растворитель отгоняли и получали 21,8 г желаемого продукта. Т.пл. 72-74оС. В дальнейшем синтез был проведен таким же образом, что и в ссылочном примере 1 для получения промежуточных соединений (3) и (4) и целевого продукта (5). 3) Метиловый эфир 3-амино-2,4-дихлорбензойной кислоты, маслянистое вещество. 4) Метиловый эфир 2,4-дихлор-3-иодобензойной кислоты, маслянистое вещество. 5) 2,4-дихлор-3-метоксикарбонилбензойная кислота, т.пл. 183-185оС. С с ы л о ч н ы й п р и м е р 12. Получение 2-хлор-3-цианометил-4-метансульфонилбен- зойной кислоты. 1) Метиловый эфир 2-хлор-3-цианометил-4-метансульфонилбензойной кислоты. 5,0 г метилового эфира 3-бромметил-2-хлор-4-метансульфонилбензойной кислоты добавляли к раствору 0,4 г 18-крауна-6 и 1,9 г цианистого калия в 50 мл ацетонитрила. Смесь перемешивали 72 ч при комнатной температуре. После отфильтровывания твердой фазы к фильтрату добавляли воду и смесь экстрагировали хлороформом. После промывки экстракта водой и сушки растворитель отгоняли и получали неочищенный продукт. Сырой продукт очищали хроматографией на короткой колонке силикагеля (элюировали хлороформом) и получали 4,1 г желаемого соединения. Т.пл. 151-155оС. 2) 2-хлор-3-цианометил-4-метансульфонилбензойная кислота. К 4,0 г метилового эфира 2-хлор-3-цианометил-4-метансульфонилбензойной кислоты и 50 мл метанола постепенно добавляли 5 мл водного раствора, содержащего 0,72 г 93% -ной гидроокиси натрия. Смесь перемешивали 15 мин при комнатной температуре. Затем реакционную смесь нейтрализовали разбавленной хлористоводородной кислотой, метанол отгоняли при пониженном давлении и концентрированный раствор экстрагировали хлороформом. После промывки экстракта водой и сушки его, хлороформ отгоняли и получали 0,9 г желаемого соединения, т.пл. 169-172оС. С с ы л о ч н ы й п р и м е р 13. Получение 3-ацетоксиметил-2-хлор-4-метансульфонил- бензойной кислоты. 1) Метиловый эфир 3-ацетоксиметил-2-хлор-4-метансульфонилбензойной кислоты. 50 мл раствора диметилформамида, содержащего 5,0 г метилового эфира 3-бромметил-2-хлор-4-метансульфонилбензойной кислоты и 1,2 г ацетата натрия, перемешивали 2 ч при 100оС. После охлаждения реакционную смесь выливали в воду с льдом и экстрагировали хлороформом. После промывки экстракта водой и сушки растворитель отгоняли и получали 4,2 г желаемого соединения. Т.пл. 165-168оС. 2) 2-хлор-3-оксиметил-4-метансульфонилбензойная кислота. 6 мл водного раствора, содержащего 1,3 г 93%-ной гидроокиси натрия, добавляли к 3,9 г метилового эфира 3-ацетоксиметил-2-хлор-4-метансульфонилбензойной кислоты в 100 мл метанола. Смесь перемешивали 30 мин при комнатной температуре. В нее добавляли 50 мл воды и метанол отгоняли при пониженном давлении. Затем реакционную смесь подкисляли хлористоводородной кислотой и экстрагировали хлороформом. Экстракт концентрировали досуха и получали 1,3 г желаемого соединения. Т.пл. 240-245оС. 3) 3-ацетоксиметил-2-хлор-4-метансульфонилбензойная кислота. 1,3 г 2-хлор-3-оксиметил-4-метансульфонилбензойной кислоты и 30 мл уксусного ангидрида нагревали с обратным холодильником 3 ч. Реакционную смесь концентрировали при пониженном давлении. Затем добавляли 50 мл воды и нагревали 1 ч. Выпавшее в осадок твердое вещество собирали фильтрацией, промывали водой и сушили. Получали 1,35 г желаемого продукта, т.пл. 219-233оС. С с ы л о ч н ы й п р и м е р 14. Получение 2,4-дихлор-3-метоксиметилбензойной кислоты. Это соединение получали таким же способом, что и в ссылочном примере 3 (1) и в примере 4. Т.пл. 130-136оС. Физические свойства промежуточных соединений следующие: 1) метиловый эфир 3-бромметил-2,4-дихлорбензойной кислоты, т.пл. 55-58оС, 2) метиловый эфир 2,4-дихлор-3-метоксиметилбензойной кислоты, маслянистое вещество. Физические свойства бензойных кислот, полученных в соответствии с предыдущими ссылочными примерами, представлены в табл.1 и 2, включающих кислоты, полученные по предшествующим ссылочным примерам. Эти бензойные кислоты легко могут быть переведены в бензоилхлориды хлорирующими средствами, такими как пятихлористый фосфор, хлористый тионил и хлористый сульфурил. При использовании таких бензойных кислот или бензоилхлоридов соединения согласно изобретению могут быть легко получены в соответствии со схемой реакций (1)-(4). Изобретение будет далее подробно описано со ссылкой на примеры. Однако следует понимать, что изобретение ни в коей мере не ограничивается такими специфическими примерами осуществления. П р и м е р 1. Получение 1-этил-5-окси-4-(4-метансульфонил-3-метоксиметил-2-ме- тилбензоил)-пиразола. 1,12 г (0,01 моля) 1-этил-5-оксипиразола растворяли в 30 мл трет-амилового спирта и в раствор последовательно добавляли 2,59 г (0,01 моля) 4-метансульфонил-3-метоксиметил-2-метилбензойной кислоты, 2,06 г (0,01 моля) N, N"-дициклогексилкарбодиимида и 0,69 г (0,005 моля) безводного карбоната калия. Реакцию в смеси проводили при температуре 80-90оС при перемешивании в течение 8 ч. После завершения реакции трет-амиловый спирт отгоняли при пониженном давлении и к остатку добавляли 30 мл воды для растворения растворимых компонентов. Смесь подвергали фильтрации для отделения нерастворимых фракций. Полученный водный раствор промывали хлороформом и добавляли концентрированную соляную кислоту, чтобы установить рН 1. Выпавший в осадок масляный компонент экстрагировали хлороформом. Растворитель отгоняли при пониженном давлении и остаток очищали хроматографией на колонке силикагеля (элюировали смесью этилацетат-этанол в отношении 9:1) и получали 2,3 г целевого продукта (выход 66% т.пл. 116-118оС). П р и м е р 2. Получение 1-этил-5-окси-4-(4-метансульфонил-3-метоксикарбонил-2-метилбензоил)-пиразола 1,12 г (0,01 моля) 1-этил-5-оксипиразола растворяли в 30 мл трет-амилового спирта и к полученному раствору последовательно добавляли 2,72 г (0,01 моля) 4-метансульфонил-3-метоксикарбонил-2-метилбензойной кислоты, 2,27 г (0,11 моля) N,N"-дициклогексилкарбонимида и 0,76 г (0,0055 моля) безводного карбоната калия. Смесь реагировала при 80оС в течение 6 ч при перемешивании. После завершения реакции трет-амиловый спирт отгоняли при пониженном давлении и затем к остатку добавляли воду для растворения растворимых компонентов. Смесь подвергали фильтрации для отделения нерастворимых веществ. Полученный водный раствор два раза промывали хлороформом и затем добавляли концентрированную хлористоводородную кислоту для установления значения рН менее 1. Выпавший в осадок масляный компонент экстрагировали хлороформом. Хлороформовый слой последовательно промывали водой и насыщенным водным раствором хлористого натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и полученный остаток перекристаллизовывали из смеси вода-этанол и получали 2,26 г целевого соединения (выход 62% т.пл. 150-152оС). П р и м е р 3. Получение 5-окси-(3-изопропоксикарбонил-4-метансульфонил-2-ме- тилбензоил)-1-метилпиразола. Операция и обработка были выполнены таким же образом, что и в примере 1, за исключением того, что 1,12 г 1-этил-5-оксипиразола заменили 0,98 г 5-окси-1-метилпиразола и 2,72 г 4-метансульфонил- 3-метоксикарбонил-2-метилбензойной кислоты были заменены 3,0 г 3-изопропоксикарбонил-4-метансульфонил-2-метилбен- зойной кислоты, и получали 1,71 г целевого соединения (выход 45% т.пл. 192-194оС). П р и м е р 4. Получение 4-(2-хлор-3-этилтиометил-4-метансульфонилбензоил)-1- этил- 5-оксипиразола. 3 г 2-хлор-3-этилтиометил-4-метансульфонилбензойной кислоты, 0,72 г карбоната калия, 50 мл трет-амилового спирта, 1,95 г N,N"-дициклогексилкарбонимида и 4,5 г 25%-ного раствора 1-этил-5-пиразола в трет-амиловом спирте смешивали и нагревали при помешивании при 70-80оС в течение 4 ч. После охлаждения смесь дистиллировали при пониженном давлении и к остатку добавляли 200 мл воды. После отфильтрования нерастворимых компонентов фильтрат промывали хлороформом. К водному слою добавляли хлористоводородную кислоту и смесь экстрагировали хлороформом. Экстракт сушили, растворитель отгоняли и получали желаемое соединение в виде неочищенного продукта. Сырой продукт перекристаллизовывали из этанола и получали 1,88 г очищенного соединения (т.пл. 142-145оС). П р и м е р 5. Получение 4-(2-хлор-5-этансульфонилметил-4-метансульфонилбензо-ил)-1-этил-5-оксипиразо ла. 0,5 г соединения, полученного в примере 4, растворяли в растворе, содержащем 30 мл хлороформа и 30 мл тетрагидрофурана при комнатной температуре и добавляли при охлаждении в ледяной бане 2,2 эквивалента м-хлорнадбензойной кислоты. Смесь постепенно нагревали до комнатной температуры и перемешивали один день. Растворитель отгоняли и полученные кристаллы собирали фильтрацией и промывали этиловым эфиром. Получали 2,2 г целевого продукта, т.пл. 133-135оС). П р и м е р 6. Получение 4-(2-хлор-3-этансульфинил-4-метансульфонилбензоил)-1- этил -5-оксипиразола. 0,45 г соединения, полученного в примере 4, растворяли в 30 мл диоксана и добавляли 0,21 г тригидрата бромистого натрия. Смесь перемешивали при комнатной температуре 30 мин и затем добавляли воду. Смесь экстрагировали хлороформом. Экстракт сушили, растворитель отгоняли и получали неочищенный продукт. Сырой продукт очищали колоночной хроматографией (элюировали смесью хлороформ-этанол) и получали 0,2 г целевого соединения в виде маслянистого вещества. П р и м е р 7. Получение 4-(2,4-дихлор-3-метоксикарбонилбензоил)-1-этил-5-оксипи- разола. Это соединение получали таким же способом, что и в примере 2. Т.пл. 167-170оС. П р и м е р 8. Получение 5-бензилокси-4-(2,4-дихлор-3-метоксикарбонилбензоил)-1-этилпиразола. Раствор, полученный растворением 0,3 г соединения, полученного в примере 7, и 0,1 г триэтиламина в 13 мл бензола, перемешивали при комнатной температуре 30 мин и затем при 50оС 3 ч. Нерастворимые вещества отфильтровывали, фильтрат концентрировали при пониженном давлении. Концентрированный продукт очищали хроматографией на колонке силикагеля (элюировали смесью бензол-этилацетат) и получали 0,15 г желаемого соединения в виде маслянистого вещества. П р и м е р 9. Получение 5-окси-4-(4-метансульфонил-3-метоксиметил-2-метилбензо- ил) -3-метоксиметил-1-метилпиразола. 1) 5-(4-метансульфонил-3-метоксиметил-2-метилбензоил)-окси-3-метоксиметил-1- метилпиразола. 1,9 г 5-окси-3-метоксиметилпиразола добавляли к смеси, содержащей 8 мл водного раствора, содержащего 0,5 г 85%-ной гидроокиси калия и 12 мл хлороформа, и затем к ней добавляли 4-метансульфонил-3-метоксиметил-2-метилбензоилхлорид. Смесь перемешивали 3 ч при комнатной температуре. Затем реакционную смесь экстрагировали хлороформом. Хлороформовый раствор промывали водой, сушили и получали желаемое соединение довольно высокого качества в виде маслянистого вещества. 2) 5-окси-4-(4-метансульфонил-3-метоксиметил-2-метилбензоил)-3- метоксиметил-1-метилпиразола. 3,0 г соединения, полученного на стадии (1), 2,7 г карбоната калия и 8 мл диоксана перемешивали при 120оС 3,5 ч. Добавляли 20 мл воды и смесь оставляли для охлаждения. Реакционную смесь промывали хлороформом и подкисляли хлористоводородной кислотой. Реакционный раствор экстрагировали хлороформом, промывали водой, сушили и получали 1,8 г неочищенного продукта. Сырой продукт перекристаллизовывали из этанола и получали 1,2 г желаемого соединения. Т.пл. 100-104оС. П р и м е р 10. Получение 4-(3-ацетоксиметил-2-хлор-4-метансульфонилбензоил)-1-этил-5-оксипиразола. Это соединение получали таким же способом, что и в примере 2. Т.пл. 140-144оС. П р и м е р 11. Получение 4-(2-хлор-3-оксиметил-4-метансульфонил-бензоил)-1-этил- 5-оксипиразола. К 30 мл метанолового раствора, содержащего 0,3 г соединения, полученного в примере 10, добавляли 5 мл водного раствора, содержащего 0,1 г 93%-ной гидроокиси натрия и смесь перемешивали 2 ч при комнатной температуре. Метанол отгоняли при пониженном давлении. Затем добавляли хлористоводородную кислоту к полученному остатку. Выпавший в осадок продукт собирали фильтрацией и получали 0,2 г желаемого соединения. Т.пл. 70-76оС. Физические свойства соединений, полученных таким же образом, что и в предыдущих примерах, представлены в табл.3 и 4, включающих и соединения, полученные в предыдущих примерах. Номер соединения в табл.3 соответствует номеру соединения в табл.4. Если соединение согласно изобретению подлежит использованию в качестве сельскохозяйственного или садового гербицида, его обычно смешивают с подходящим носителем, например с твердым носителем, таким как глина, тальк, бентонит или диатомовая земля, или с жидким носителем, таким как вода, спирт (например, метанол или этанол), ароматический углеводород (например, бензол, толуол или ксилол), хлорированный углеводород, простой эфир, кетон, сложный эфир (например, этилацетат) или амид кислоты (например, диметилформамид). Если желают получить оптимальную композицию, такую как жидкий препарат, эмульгирующийся концентрат, смачивающийся порошок, дуст, гранула или суспензия, к соединению согласно изобретению и носителю может быть добавлен эмульгатор, диспергатор, суспендирующее средство, средство, способствующее проникновению и распределению (растеканию) композиции, или стабилизатор. Далее, если необходимо, то добавляют другие гербициды, различные инсектициды, бактерициды, регуляторы роста растений или синергические средства в композицию в процессе ее приготовления или во время применения гербицида. Другие гербициды могут быть совмещены с гербицидом согласно изобретению, например, соединения, описанные в Farm Chemicals Hondbook, the 73 rd Edition, 1987. Среди них могут быть упомянуты, например, атразин, цианазин, алахлор, метолахлор, ЕРТС, 2,4-Д, бутилат, дикамба, бромоксинил и тридифан. Далее могут быть также объединены с гербицидом согласно настоящему изобретению N-/(4,6-диметоксипиримидин-2-ил)-аминокарбонил/-3-хлор -4- метоксикарбонил-1-метилпиразол-5-суль-фонамид или N-/(4,6-диметоксипиримидин-2-ил)-аминокарбонил)-3-бром-4- метоксикарбонил-1-метилпиразол-5-сульфонамид, описанные в патенте США N 4668277. Доза варьирует в зависимости от места применения, сезона, метода применения, вида культурного растения и тому подобного. В общем доза в пределах 0,001-10 кг на 1 га активного ингредиента обычно является приемлемой. Далее следуют примеры композиций, содержащих в качестве гербицида соединения согласно изобретению, как активный ингредиент. Следует понимать, что настоящее изобретение ни в коей мере не ограничивается этими конкретными примерами. П р и м е р к о м п о з и ц и и 1. Смачивающийся порошок, мас.ч. Соединение N 3 согласно изобретению 60 Цееклит РFР (торговое название глины каолино- вого типа, производство Zuklite Industries Co, Ltd 33 Cорпол 5039 (торговое название смеси неионоген- ного и анионоактивного поверхностно-активного вещества, производимого Toho Chemical, Co Ltd 5 Карплекс (торговое название средства, предотвращающего коагуляцию, состоящего из смеси поверхностно- активного вещества и тонкодисперсного порошка кремнезема, производство Shionogi Pharmaceutical Co. Ltd 2 Перечисленные компоненты гомогенно измельчают в порошок и смешивают для получения смачивающегося порошка. П р и м е р к о м п о з и ц и и 2. Смачивающийся порошок, мас.ч. Соединение N 7 согласно изобретению 60 Цееклит РFР (торговое название глины каолинового типа, выпускаемой Zuklite Industries Co. Ltd 33 Cорпол 5039 (торговое название смеси неионогенного и анионоактивного поверхностно-активного вещества, производство Toho Chemical Cо. Ltd 5 Карплекс (торговое название средства, предотвращающего коагуляцию, образованного смесью поверхностно-активного вещества и тонкодисперсного порошка кремнезема, производимого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 3. Смачивающийся порошок, мас.ч. Соединение N 15 согласно изобретению 60 Цееклит РFР (торговое название глины каолинового типа, производимой Zuklite Industries Co. Ltd 33 Сорпол 5039 (торговое название смеси неионогенного и анионоактивного поверхностно-активного вещества, производимой Toho Chemical Co. Ltd 5 Карплекс (торговое название средства, предотвращающего коагуляцию, состоящее из смеси поверхностно-активного вещества и тонкодисперсного порошка кремнезема, производимого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 4. Смачивающийся порошок, мас.ч. Соединение согласно изобретению N 21 60 Цееклит РFР (торговое название глины каолинового типа, производство Zuklite Industries Co. Ltd 33 Cорпол 5039 (торговое название смеси неионоген- ного и анионоактивного поверхностно-активного вещества, производство Toho Chemical Co. Ltd 5 Карплекс (торговое название средства, предотвращающего коагуляцию, являющегося смесью поверхностно- активного вещества и тонко- дисперсного порошка кремне- зема, выпускаемого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 5. Смачивающийся порошок, мас.ч. Соединение N 25 согласно изобретению 60 Цееклит РFP (торговое название глины каолинового типа, выпускаемой Luklite Industries Co. Ltd 33 Cорпол 5039 (торговое название смеси неионоген- ного и анионоактивного поверхностно-активного ве- щества, выпускаемого Toho Chemical Co. Ltd 5 Карплекс (торговое название средства, предотвраща- ющего коагуляцию, состоящего из смеси поверхностно- активного вещества и тонкодисперсного порошка кремнезема, выпускаемого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 6. Смачивающийся порошок, мас.ч. Соединение N 35 согласно изобретению 60 Цееклит РFР (торговое название глины каолинового типа, выпускаемой Zuklite Industries Co. Ltd 33 Cорпол 5039 (торговое название смеси неионоген- ного и анионоактивного поверхносно-активного вещества, производимого Toho Chemical Co. Ltd 5 Карплекс (торговое название антикоагулянта, являющегося смесью поверхностно-активного вещества и тонкодисперсного порошка кремнезема, изготавливаемого Shionogi Pharmaceutical Co. Ltd 2 П р и м е р к о м п о з и ц и и 7. Эмульгирующийся концентрат, мас.ч. Соединение N 3 согласно изобретению 1,5 Ксилол 78,6 N,N-диметилформамид 15 Сорпол 2680 (торговое название смеси неионо- генного и анионоактивного поверхностно-активного вещества, выпускаемого Togo Chemical Co. Ltd 5 Перечисленные выше ингредиенты гомогенно смешивали для получения эмульгирующего концентрата. П р и м е р к о м п о з и ц и и 8. Эмульгирующийся концентрат, мас.ч. Соединение N 11 согласно изобретению 1,5 Ксилол 78,5 N,N-диметилформамид 15 Сорпол 2680 (торговое название смеси неионогенного и анионо- активного поверхностно- активного вещества, производимого 5 П р и м е р к о м п о з и ц и и 9. Суспензия, мас.ч. Соединение N 3 согласно изобретению 40 Агризол В-710 (торговое название неионогенного поверхностно-активного вещества, выпускаемого Kao Corporation 10 Рунокс 1000 С (торговое название анионоактив- ного поверхностно- активного вещества, выпуска- емого Toho Chemical Co. Ltd 0,5 1% Родопол-вода (торговое название загустителя, выпускаемого Rhone-Paulenc 20 Вода 29,5 Перечисленные ингредиенты гомогенно смешивали для получения суспензии. П р и м е р к о м п о з и ц и и 10. Суспензия, мас.ч. Соединение N 10 согласно изобретению 40 Агризол В-710 (торговое название неионогенного поверхностно-активного вещества, выпускаемого Као Cоrporation 10 Рунокс 1000 С (торговое название неионоактивного поверхностно-активного вещества, выпускаемого Toho Chemical Co. Ltd 0,5 1% Родопол-вода (торговое название загустителя, выпускаемого Rhone Poulenс 20 Вода 29,5 П р и м е р к о м п о з и ц и и 11. Жидкая композиция, мас.ч. Соединение N 39 согласно изобретению 30 Ниппол (торговое название неионогенного поверхностно-активного вещества, выпускаемого Nissan Chemical Industries, Ltd 10 Вода 60 Перечисленные выше компоненты гомогенно смешивали для получения жидкой композиции. П р и м е р к о м п о з и ц и и 12. Жидкая композиция, мас.ч. Соединение N 40 согласно изобретению 30 Ниппол (торговое название неионогенного поверхностно-активного вещества, производимого Nissan Chemical Indusctries, Ltd 10 Вода 60 П р и м е р к о м п о з и ц и и 13. Жидкая форма, мас.ч. Соединение N 46 согласно изобретению 30 Ниппол (торговое название неионогенного поверхностно-активного вещества, выпускаемого Nissan Chemical Industries, Ltd 10 Вода 60 П р и м е р к о м п о з и ц и и 14. Жидкая форма, мас.ч. Соединение N 41 согласно изобретению 10 Сорпол W-150 (торговое название неионогенно- го поверхностно-активного вещества, производимого Toho Chemical Co. Ltd 10 Вода 80 Перечисленные выше ингредиенты гомогенно смешивали для получения жидкой формы препарата. При использовании упомянутые выше смачивающиеся порошки, эмульгирующиеся концентраты, суспензии и жидкие формы композиций разбавляют водой в 50-1000 раз и применяют в количествах, обеспечивающих 0,001-5 кг активного ингредиента на 1 га. Соединения согласно изобретению применимы не только на сельскохозяйственных и садовых плантациях, таких как суходольные поля, рисовые поля и фруктовые сады, но и на площадях, изъятых из сельскохозяйственного употребления, таких как стадионы, пустоши и полосы отчуждения вдоль железных дорог для борьбы с различными сорняками. Доза при их применении варьирует в зависимости от применяемого метода, обрабатываемой площади, сезона применения, вида культурных растений и так далее. Однако обычно она находится в пределах 0,001-5,0 кг на 1 га. Далее гербицидные активности соединений согласно изобретению будут описаны со ссылкой на конкретные примеры испытаний. П р и м е р и с п ы т а н и й 1. Тест на гербицидное действие при обработке почвы. Пластиковый ящик длиной 22 см, шириной 15 см и глубиной 6 см заполняли дилювиальной почвой, подвергающейся стерилизации, и в нее высевали семена Echinochloa crus-galli, Setaria viridis, Eleusine indica, Digitaria adscendens, Panicum dichotomiflorum, Abutilon theophrasti, Amaranthus lividus, Polygonum longisetum и Zea maus и высаживали клубни Сyperus esculentus. Семена засыпали слоем почвы толщиной около 1,5 см и затем на поверхность почвы равномерно наносили раствор гербицида, обеспечивающий заданную концентрацию активного ингредиента. Гербицидный раствор готовили разбавлением водой смачивающегося порошка, эмульгирующегося концентрата, жидкой композиции или суспензии и наносили с помощью ручного распылителя на всю поверхность почвы. Через три недели после применения гербицидного раствора определяли гербицидное действие против каждого вида сорняков на основе следующих стандартных оценок: полученные результаты представлены в табл.5. Номер соединения в табл.5 соответствует номеру соединения в табл.3. Стандартные оценки: 5 степень подавления роста более 90% (почти полное увядание) 4 степень подавления роста 70-90% 3 степень подавления роста 40-70% 2 степень подавления роста 20-40% 1 степень подавления роста 5-20% 0 степень подавления роста менее 5% (почти неэффективно). Упомянутые оценки подавления роста рассчитывали по следующему уравнению: степень подавления роста (%) (1 Т/N) х 100, где Т вес зеленой массы над поверхностью почвы обработанного участка; N зеленой массы над поверхностью почвы необработанного участка. П р и м е р и с п ы т а н и й 2. Тест на гербицидное действие при обработке листьев. Пластиковые ящики длиной 22 см, шириной 15 см и глубиной 6 см заполняли стерилизованной дилювиальной почвой и семена Echinochloa crus-galli, Setaria viridis, Eleusine indica, Digitaria adscendens, Panicum dichotomiflorum, Xanthium strumarium, abutilon theophrasti, Amaranthus lividus, Polygonum longisetum и Zea mays высевали местами и высаживали клубни Сyperus esculentus. Затем семена покрывали слоем почвы толщиной около 1,5 см. Сорняки и культурные растения выращивали до стадии 2 или 3 листа и гербицидный раствор равномерно разбрызгивали на листья так, чтобы активный ингредиент был применен в заранее заданной концентрации. Гербицидный раствор готовили разбавлением смачивающегося порошка, эмульгирующегося концентрата, жидкой формы композиции или суспензии, описанных в примерах получения композиций, водой и наносили на всю поверхность листьев сорняков и культурных растений с помощью небольшого распылителя. Через две недели после применения гербицидного раствора определяли гербицидное действие против каждого вида сорняков на основе стандартных оценок, описанных в примере испытаний 1, и фитотоксичность против каждого вида культурных растений определяли также на основе стандартных оценок, приведенных в примере испытаний 1. Результаты представлены в табл.6. Номер соединений в табл. 6 соответствует номеру соединения в табл.3. В табл. 5 и 6 применены следующие сокращения: доза доза активного ингредиента (г/ар) ЕС Echinochloa crus-galli (просо куриное) SE Setaria viridis (щетинник зеленый) EL Eleusine indica (элеузина индийская) DI Digitaria adscendens (росичка кровяная) РА Panicum dichotomiflorum (просо) АВ Abutilon theophrasti (канатник Теофраста) АМ Amaranthus lividus (ширица синеватая) PO Polygonum longisetum (горец длинностолбиковый) ХА Kanthum strumarium (дурнишник) СУ Сyperus esculentus (сыть съедобная) ZE Zea mays (кукуруза) 2,47 (3Н, с), 3,22 (3Н,с), 3,50 (3Н,с), 3,69 (3Н,с), 4,96 (2Н,с), 7,30 (1Н,с), 7,78 (2Н, А-В кв), 10,9 (1Н) /СDCl3/ 2,41 (3Н,с), 3,17 (3Н,с), 3,50 (3Н,с), 4,94 (2Н,с), 5,53 (2Н,с), 7,30-8,12 (8Н,м) /CDCl3) 1,44 (3Н, т), 2,48 (3Н,с), 3,23 (3Н,с), 3,51 (3Н,с), 4,07 (2Н,кв), 4,98 (2Н,с), 7,36 (1Н,с), 7,82 (2Н, А-В кв) /CDCl3/ 1,48 (6Н, д), 2,47 (3Н,с), 3,19 (3Н,с), 3,48 (3Н,с), 4,53 (1Н,м), 4,92 (2Н,с), 7,18 (1Н,с), 7,69 (2Н, А-В кв), 9,57 (1Н) /CDCl3/ 2,43 (3Н,с), 3,78 (3Н,с), 3,94 (3Н,с), 7,24-7,81 (7Н,м), /CDCl3/ 1,07 (3Н, т), 2,27 (3Н,с), 3,01 (3Н,с), 3,30-3,65 (5Н,м), 4,77 (2Н,с), 6,99 (1Н,с), 7,48 (А-В кв), 8,22 (1Н,с) /CDCl3/ 1,45 (3Н,т), 3,04 (2Н,кв), 4,05 (2Н,кв), 4,91 (2Н,кв), 7,29 (1Н,с), 7,70 (1Н,с), 7,85 (2Н,кв) (CDCl3) 0,91-2,03 (8Н,м), 2,41 (3Н,с), 3,20 (3Н,с), 3,91-4,47 (4Н,м), 7,36-8,08 (4Н,м), (CDCl3). 1,26 (3Н, т), 1,49 (3Н,д), 2,49 (3Н,с), 3,24 (3Н,с) 3,69 (2Н,кв), 4,59 (1Н,м), 5,00 (2Н,с), 7,28-8,14 (4Н,м), /CDCl3/ 1,27 (3Н, т), 1,67 (3Н,с), 2,42 (3Н,с), 3,23 (3Н,с), 3,63 (3Н,с), 3,68 (2Н,кв), 5,02 (2Н,с), 7,7 (1Н,с), 7,75 (2Н, А-В кв), (СDCl3) 1,45 (3Н,т), 2,49 (шир.с), 3,44 (3Н,с), 4,03 (2Н,кв), 4,66 (2Н,с), 7,29 (2Н,кв.), 7,39 (1Н,с); 1,47 (3Н, т), 3,29 (3Н,с), 3,51 (3Н,с),3,82 (3Н,с), 4,09 (2Н,кв), 5,00 (2Н,с), 7,50 (1Н,с), 7,84 (2Н, А-В кв), 7,96 (1Н, с), /CDCl3/ 1,07-1,67 (9Н, м), 2,59 (3Н,с), 3,17 (3Н,с), 3,42 (2Н,т), 3,96 (2Н,т), 5,61 (1Н,кв), 7,27 (1Н,с), 7,61 (2Н,А-В кв), 9,66 (1Н, широкий с) /CDCl3/ 1,09 (3Н, т), 1,45 (3Н,т), 2,55 (3Н,с), 3,16 (3Н,с) 3,27 (3Н,с), 3,70-4,20 (4Н, м), 5,13-5,36 (1Н, м) 7,14 (1Н,с), 7,60 (2Н, А-В кв), 9,46 (1Н, широкий с) /СDCl3/
ФОРМУЛА ИЗОБРЕТЕНИЯПроизводные пиразола общей формулы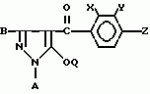 где A - С1-С3-алкил; B - водород или С1-С3-алкил; X - С1-С6-алкил, хлор или С1-С6-алкокси; Y - группа общей формулы COOR1, где R1 - водород, С1-С6-алкил, хлор, C1-C6-алкил или метоксиэтил, или группа общей формулы ZOR2, где Z - метилен, возможно замещенный метилом или этилом, R2 - водород, С1-С8-алкил, 2,2,2-трифторэтил или пропаргил, или группа общей формулы CH2-O-CH2-CH2-OR3, где R3 - С1-С6-алкил, или группа общей формулы CH2R4, где R4 - циано или S(O)nR5, где R5 - С1-С6-алкил, n = 0-2-целое число, или группа общей формулы CH = CHOR6, где R6 - С1-С6-алкил; Z - хлор или метилсульфонил; Q - водород, фенилсульфонил или бензил, или их соли с основаниями. Приоритет по признакам: 17.03.87 при A-этил, B-водород, X-метил, Y-группа CH2OCH3, Z-метилсульфонил, Q-водород. при 17.07.87 при A-С1-С3-алкил, B-водород, X-С1-С6-алкил, Y-группа COOR1, где R1-С1-С6-алкил, Z-метилсульфонил, Q - водород. 30.09.87 при A - С1-С6-алкил, B-водород, С1-С3-алкил, X-C1-C6-алкил, хлор С1-С6-алкокси, Y - группа COOR1, где R1 - водород, С1-С6-алкил, метоксиэтил, или группа ZOR2, где Z - метилен, возможно замещенный метилом, R2 - С1-С8-алкил, пропаргил, или группа CH2OCH2 CH2OR3, где R3 - С1-С6-алкил, или группа CH2R4, где R4 = S(O)nR5, где n = 0 - 2, R5 - С1-С6-алкил, Z-хлор или метилсульфонил, Q - водород, бензил, фенилсульфонил, соли с основаниями. 13.01.88 при A - С1-С6-алкил, B-водород, X - С1-С6-алкил, хлор, С1-С6-алкокси, Y-группа COOR1, где R1 - С1-С6-алкил, хлор, С1-С6-алкил, или группа ZOR2, где R2 - водород, С1-С8-алкил, 2,2,2-трифторэтил, или группа CH2R4, R4-циано, или группа CH= CHR6, где R6 - С1-С6-алкил, Z - хлор или метилсульфонил, Q - водород, бензил, соли с основаниями. где A - С1-С3-алкил; B - водород или С1-С3-алкил; X - С1-С6-алкил, хлор или С1-С6-алкокси; Y - группа общей формулы COOR1, где R1 - водород, С1-С6-алкил, хлор, C1-C6-алкил или метоксиэтил, или группа общей формулы ZOR2, где Z - метилен, возможно замещенный метилом или этилом, R2 - водород, С1-С8-алкил, 2,2,2-трифторэтил или пропаргил, или группа общей формулы CH2-O-CH2-CH2-OR3, где R3 - С1-С6-алкил, или группа общей формулы CH2R4, где R4 - циано или S(O)nR5, где R5 - С1-С6-алкил, n = 0-2-целое число, или группа общей формулы CH = CHOR6, где R6 - С1-С6-алкил; Z - хлор или метилсульфонил; Q - водород, фенилсульфонил или бензил, или их соли с основаниями. Приоритет по признакам: 17.03.87 при A-этил, B-водород, X-метил, Y-группа CH2OCH3, Z-метилсульфонил, Q-водород. при 17.07.87 при A-С1-С3-алкил, B-водород, X-С1-С6-алкил, Y-группа COOR1, где R1-С1-С6-алкил, Z-метилсульфонил, Q - водород. 30.09.87 при A - С1-С6-алкил, B-водород, С1-С3-алкил, X-C1-C6-алкил, хлор С1-С6-алкокси, Y - группа COOR1, где R1 - водород, С1-С6-алкил, метоксиэтил, или группа ZOR2, где Z - метилен, возможно замещенный метилом, R2 - С1-С8-алкил, пропаргил, или группа CH2OCH2 CH2OR3, где R3 - С1-С6-алкил, или группа CH2R4, где R4 = S(O)nR5, где n = 0 - 2, R5 - С1-С6-алкил, Z-хлор или метилсульфонил, Q - водород, бензил, фенилсульфонил, соли с основаниями. 13.01.88 при A - С1-С6-алкил, B-водород, X - С1-С6-алкил, хлор, С1-С6-алкокси, Y-группа COOR1, где R1 - С1-С6-алкил, хлор, С1-С6-алкил, или группа ZOR2, где R2 - водород, С1-С8-алкил, 2,2,2-трифторэтил, или группа CH2R4, R4-циано, или группа CH= CHR6, где R6 - С1-С6-алкил, Z - хлор или метилсульфонил, Q - водород, бензил, соли с основаниями.Популярные патенты: 2420949 Способ оценки потенциальной урожайности семянок сафлора красильного ... соцветиях, бобах отличается не существенно. Таким образом, описанный способ не приемлем для оценки потенциальной продуктивности масличных культур при возделывании в иных почвенно-климатических условиях.Известен, кроме описанных, способ оценки потенциальной продуктивности сельскохозяйственных растений, включающий отбор пробы листьев, измерение биофизических показателей, характеризующих сочетание двух фитосистем и оценку потенциальной продуктивности по значению измеренных показателей, в котором, с целью повышения достоверности способа, отбор пробы осуществляют при освещенности интенсивностью 10-40 Вт/м2 , непосредственно после отбора пробу погружают в жидкий азот, измерение ... 2199860 Способ увеличения устойчивости подсолнечника к действию гербицида ... ... 2111642 Высевающий аппарат ... показателей работы высевающего аппарата в целом. Кроме того, при отключении высевающего устройства возможно травмирование семян между кромками неподвижного и подвижного колец. Цель изобретения - повышение надежности работы, снижение травмирования семян и неравномерности высева через отдельные высевные отверстия высевающего устройства независимо от рельефа засеваемого поля. Указанная цель достигается тем, что в днище высевающего устройства прямоугольной формы в два ряда размещены высевные продолговатые отверстия, осевые линии которых совпадают с направлением колебаний корпуса высевающего устройства, сверху на днище его устанавливается и закрепляется винтами через продолговатые ... 2218756 Способ изготовления антипаразитарного ошейника ... режимах в материальном цилиндре экструдера: зона загрузки - 40oС, зона пластикации - 65oС, зона дозирования - 85oС, формующая головка - 85oС. В заявленном способе в качестве термопластичного полимера используют сэвилен - продукт совместной полимеризации этилена с винилацетатом в массе под высоким давлением. Сополимер этилена и винилацетата обладает высокой эластичностью и большей стойкостью к окислению, чем поливинилхлорид, не токсичен, не требует специальной пластификации, разрешен для использования в медицине. Заявленный способ изготовления антипаразитарного ошейника по сравнению с известными аналогичными способами позволяет повысить эффективность способа благодаря ... 2228022 Способ ведения виноградных кустов ... пасынков второго порядка. Кусты приняли вид средней чашевидной формы.В течение третьей и последующих вегетаций побеги, развившиеся на сучках замещения и плодовых стрелках, свободно размещались вокруг сплетенного штамба куста. Нагрузка в среднем на куст составила: в глазках - 25-30 шт, в побегах - 20-25 шт. Большинство побегов были плодоносные. Кусты вступили в первое плодоношение на третий год, а в период полного плодоношения - на четвертый год жизни.На контроле формирование и обрезка виноградных кустов осуществлялась по технологии, принятой в качестве прототипа.Весной второго года после посадки кусты были обрезаны с оставлением на каждом по одному сучку с двумя глазками. Вдоль оси ... |
Еще из этого раздела: 2472951 Машина (варианты) 2440721 Способ определения вредоносности насекомых комплекса "гнус" для крупного рогатого скота 2460269 Малогабаритный картофелеуборочный комбайн 2056100 Доильный стакан 2112361 Контроллер программируемого управления поливом 2195102 Устройство для отделения грунта и земли от корней и корневищ солодки в качестве лакричного сырья 2193304 Захват лесозаготовительной машины 2102853 Питательное устройство для растений 2440712 Автоматизированная система для хранения в поле, возможности оперативного контроля и выгрузки убранных продуктов урожая из уборочной машины 2160520 Способ создания лакричных плантаций, предпочтительно солодки голой, на бросовых землях |
Изобретения в сельском хозяйстве
Обработка почвы в сельском и лесном хозяйствах
Посадка, посев, удобрение
Уборка урожая, жатва
Обработка и хранение продуктов полеводства и садоводства
Садоводство, разведение овощей, цветов, риса, фруктов, винограда, лесное хозяйство
Новые виды растений или способы их выращивания
Производство молочных продуктов
Животноводство, разведение и содержание птицы, рыбы, насекомых, рыбоводство, рыболовство
Поимка, отлов или отпугивание животных
Консервирование туш животных, или растений или их частей
Биоцидная, репеллентная, аттрактантная или регулирующая рост растений активность химических соединений или препаратов
Хлебопекарные печи, машины и прочее оборудование для хлебопечения
Машины или оборудование для приготовления или обработки теста
Обработка муки или теста для выпечки, способы выпечки, мучные изделия
|
|
||